7. Decentralized Clinical Trials
- 31 Mins to read
- DarkLight
7. Decentralized Clinical Trials
- 31 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
7.1 Do the country regulations allow Decentralized Clinical Trials (DCT) elements (e.g., eConsent, ePRO administration, remote investigator site, etc.)?
Yes. There is no regulation preventing the conduct of DCTs. DCTs are allowable, subject to RA and EC approval.
7.2 Is there any specific regulation/guidance on the use of DCT elements within a clinical trial?
Yes. Clinical Research Malaysia released a guidance document regarding Decentralized Clinical Trials in July 2023.
7.3 What is the overall acceptability of DCT elements by the regulatory authorities and Ethics Committees?
The guidance states that many different aspects of decentralization will be considered as long as the overall standards of the trial continue to meet the Malaysian regulatory requirements and the safety of the patient is not compromised. Decentralized elements of a trial should be outlined in the trial documentation and are subject to ethics approval.
7.4 Has a fully virtual trial (DCT) been conducted in the country yet? If so, please provide an example including the registration number and any link to it, whether COVID/non-COVID trials.
No, there is no evidence of a fully decentralized trial being conducted in Malaysia.
7.5 Are there any non-regulatory DCT initiatives in the country, such as where the investigator sites and local CROs founded an alliance?
No.
7.6 Are there any general considerations when using DCT elements in a study?
The considerations for applying decentralized elements to a clinical trial are outlined in the guidance issued by Clinical Research Malaysia in July 2023. This guidance speaks specifically to considerations such as the use of technology for gathering trial data, gaining consent, etc.
7.7 Considerations/Requirements for the Use of eConsent/Remote Consent
e-Consent is permitted, subject to conditions as defined in the Clinical Research Malaysia guidance and summarized below:
INFORMED CONSENT PROCESS
Definition and considerations
According to Malaysian GCP, Informed Consent is a process by which a subject voluntarily confirms his or her willingness to participate in a particular trial, after having been informed of all aspects of the trial that are relevant to the subject's decision to participate. Informed consent is documented by means of a written, signed, and dated informed consent form. The relevant documentation should be protected from any unauthorized modification.
The informed consent process should be carried out in compliance with the principles laid down in the Malaysian GCP, PDPA Act, and relevant national regulations. This is applicable to both traditional methods of consenting and remote/electronic consenting (partial or full). This guidance focuses on the following methods of the consenting process:
• The use of different types of media to enhance the trial participant’s comprehension of the trial.
• The use of any electronic media (such as text, graphics, audio, video, podcasts, or websites) to convey information related to the study, and/or
• To seek and/or document informed consent via an electronic device such as a smartphone, tablet, or computer.
• Alternative methods for the electronic provision of information should be available.
The sponsor is responsible for verifying whether a clinical trial site agrees to the use and storage of electronic methods for the consent process. It should be ensured that the information provided to the trial participant is in a form that can be stored and retrieved by the trial participant.
Consenting process (in person and remotely)
In general, the consenting process should be a physical meeting between the investigator and the potential trial participant. The following should be considered in the case of adopting e-consent, be it in-person or remotely:
1. Ensure that the e-consent process is user-friendly and easily accessible for participants:
• The e-consent process should be designed to be user-friendly, intuitive, and easy to navigate for participants. Participants should be able to access the e-consent process using commonly available devices and internet browsers.
• The e-consent process should also be compatible with different types of devices, such as desktop computers, laptops, tablets, and smartphones.
2. Provide clear and concise information about the study:
• The e-consent process should provide clear and concise information about the study, including the purpose, risks, benefits, and confidentiality measures.
• The information should be presented in a language and format that participants can understand. It should also be organized in a logical and easy-to-read manner.
3. Include an option for participants to ask questions and clarify any concerns before providing e-consent (Malaysia DCT Guidance Document 10):
• The e-consent process should provide participants with the option to ask questions and clarify any concerns before providing e-consent.
• The contact details of the site study team or a helpline number should be provided for this purpose. Participants should be able to contact the study team (e.g. investigator or study coordinators) by phone, email, or online chat.
4. Verify the participants' identity before allowing them to provide e-consent:
• Added measures should be taken to verify the identity of the trial participant before they provide e-consent. This can be done through various methods, such as using a unique login ID or sending a verification code to the participant's email or phone number.
5. Ensure that the e-consent process is secure and complies with relevant data protection and privacy laws and regulations:
• The e-consent process should be designed to ensure the security and privacy of participants' personal data. It should comply with relevant data protection laws and regulations, such as the Malaysian GCP, PDPA Act, and all relevant national regulations.
• The e-consent process should use encryption and other security measures to protect the transmission and storage of personal data.
• Trial participant identifiers should not be made available to any parties outside its intended purposes. All efforts should be made to limit unauthorized access or disclosure of this information.
6. Provide participants with the option to withdraw their e-consent at any time and explain how to do so:
• The e-consent process should provide participants with the option to withdraw their e-consent at any time. This should be explained clearly in the e-consent form, and participants should be informed about the implications of withdrawing their consent.
7. Keep a record of the documentation of the e-consent process and store it securely for the duration of the trial:
• The e-consent process should be documented and stored securely for the duration of the trial. This includes the e-consent form and any other relevant documentation.
• The record of the e-consent process should be easily retrievable and auditable in case of an inspection or audit.
8. Provide clear instructions and guidance for participants on how to complete the e-consent process and provide additional resources and support when needed:
• The e-consent process should provide clear instructions and guidance for participants on how to complete the e-consent process. This includes explaining how to access and navigate the e-consent form, how to provide e-consent, how to revise their personal information in e-consent, and how to withdraw e-consent if desired.
• The instructions and guidance should be provided in a clear and concise manner, and in a language and format that participants can understand. Malaysia DCT Guidance Document 11
• Consider providing additional resources and support for participants who may have difficulty with the e-consent process.
• The e-consent process should be designed to be accessible for all participants, including those who may have difficulty with the e-consent process. This includes participants who have limited technology access, language barriers, or cognitive or visual impairments.
9. Obtain IRB/ IEC approvals/favorable opinion for the e-consent process:
• Before implementing the e-consent process, the study team should obtain IRB/ IEC approvals/favorable opinion for the e-consent process.
• The e-consent process should be designed to comply with the relevant guidelines and regulations, such as the Malaysian GCP guidelines and other relevant regulations/ guidance.
10. The e-consent process should be clearly described in the study protocol and/or other relevant documentation. Any changes or modifications to the e-consent process should be properly documented and communicated to the relevant stakeholders.
11. In situations where literacy is a barrier, alternative methods should be considered to ensure that informed consent is obtained in a meaningful and understandable manner. These methods may include using visual aids, pictures, videos, or oral presentations, depending on the cultural and linguistic context of the participants. The informed consent process should be reviewed and approved by the respective IRB/ IEC. The informed consent process or measures should be described in the protocol or relevant study document, with documented approval/favorable opinion by IRB/ IEC to protect the rights, safety, and wellbeing of the subject.
7.8 Considerations/Requirements for the Use of eSignatures
The use of e-signatures is described in the Clinical Research Malaysia Guidance and states that:
Signature (eSignature, print to sign)
- There are various ways of obtaining a signed informed consent but not limited to form by remote means. This includes:
- A hardcopy consent form sent to the participant, signed with a ‘wet ink signature’ and sent back by post, or
- An e-consent form signed with an electronic signature, i.e., completely electronic.
- The sponsor should ensure that the systems used have proportionate security levels and that safeguards regarding confidentiality are in place. In general, the electronic signature functionality should be in accordance with the requirements described in the EMA Guideline on Computerized Systems and Electronic Data in Clinical Trials.
- The method used to document the informed consent process should follow national and institutional requirements with regard to the acceptability of electronic signatures.
- eSignature practices shall comply with the Malaysia Electronic Commerce Act 2006 (ECA). Malaysia DCT Guidance Document 12
- Procedures should be in place to handle follow-up steps after the consent has been withdrawn electronically, including partial withdrawal and complete withdrawal, due to the impact on trial participant participation and data collection.
- These procedures should include timely notification to the investigator and a communication plan with all other stakeholders. By any means, withdrawals should also be possible outside of the system, and this should be recorded by the investigator.
7.9 Considerations/Requirements for Electronic Patient Reported Outcome (ePRO)
The use of electronic patient-reported outcomes (ePRO) is permitted.
The Clinical Research Malaysia guidance contained the following considerations for implementation:
When using electronic system(s) for direct data collection, the sponsor should ensure and implement adequate oversight and measures including, but not limited to:
• Ensure that all parties involved in the clinical trial have an overview of the data flow; a data flow diagram with additional explanations in the protocol is highly recommended.
• Ensure that the used data collection tools are created in a controlled manner, configured, and validated in accordance with their intended use. Appropriate change control and ongoing validation are needed.
• Determine the type and scope of the trial participants’ personal data to be collected and ensure adequate protection in compliance with the relevant Malaysian regulations.
• Ensure that when source data captured by a data collection tool is transferred to another location and subsequently deleted from the data collection tool, both the data and the metadata are transferred (refer to Malaysian GCP).
• Implement measures such as encryption to minimize the risk of unauthorized access, when transferring the data from a data collection tool to a server. Malaysia DCT Guidance Document 23
• Ensure access to trial data is controlled by defined user rights and methods of access for all relevant parties involved. Unauthorized access should be prevented using appropriate security measures (e.g., firewalls).
• Ensure control of and continuous and complete access by the investigator to both source data generated either on-site or off-site as well as source data reported to the sponsor (e.g., central laboratory data).
• Appropriate measures and procedures should be in place to minimize the risk of erroneous data entry for data measured and entered directly by trial participants, especially on primary, key-secondary, or safety endpoints. In summary, when electronic systems are used as a mode of data collection, the principles of data collection, handling, and storage requirements as stated in the Malaysian GCP and applicable regulatory requirements must be adhered to. In addition, the data protection requirements are according to the relevant Malaysia regulations, including data privacy and cybersecurity laws and regulations.
7.10 Considerations/Requirements for Home Health Care (HHC) - Home Nursing
The Clinical Research Malaysia Guidance makes the following statements in relation to home healthcare:
Home Health Care
In a clinical trial with decentralized elements, the trial-related procedures may take place outside the trial site. This makes it easier for trial subjects to participate in clinical trials by reducing the overall burden and need to travel to trial sites, promoting subject compliance and retention, and enabling subjects normally unable to participate in life-saving clinical trials.
Investigator-delegated Home Health Care Professionals (HHCPs) (e.g., nurses, phlebotomists) could now perform these used-to-be site procedures at the subject’s home. Below are the activities that can be performed by HHCPs at home, as required under the study protocol and laboratory manual, while subjected to investigator’s discretion and applicable regulations and guidance:
• Blood draws and processing (e.g., Pharmacokinetics, safety laboratory samples)
• Collection of biological samples (e.g., oral mucosal swabs, urine, fecal samples)
• Training and education of the trial subject and caretaker (e.g., IP self-administration)
• Administration of the Investigational Product (GMP, GCP)
• IP compliance check
• Clinical assessments [e.g., vital signs, concomitant medication checks, spirometry test, electrocardiograms (ECG by nurse only)]
Equipment importation, validation for portability, and calibration should also be considered for any specialized equipment used for home care visits in accordance with the regulation or requirement by the Medical Device Authority (MDA). Considerations for implementation are not limited to the following:
1) Sponsor
• Risk assessment related to home health care should be documented in the study protocol or a separate document. The trial subjects should not be exposed to higher risks than those foreseen for the same procedure applied in a clinical trial site. These risks should be conveyed to the Investigator via an Investigator Brochure/written document so that the Investigator can make an informed decision on whether a home visit is suitable.
• The procedures performed at the trial subject’s home should clearly be described in the study protocol and related informed consent form and approved by IRB/IEC and NPRA if for IP Administration for higher risk profile IP. Any exportation of human tissue samples of the trial subjects should also follow and comply with the same exportation requirements and regulations.
• Only healthcare vendors contracted by the Sponsor, and registered in Malaysia, could perform Home Health Care services per the scoped activities. Malaysia DCT Guidance Document 18
• There should be a written agreement (i.e., Clinical Trials Agreement (CTA) or additional addendum document) stating the scoped home care services, the health care vendor providing the services between the Sponsor and the Investigator/Site and relevant responsibilities and accountability by each party.
• Professional liability/ professional indemnity/ clinical trial insurance should be extended to cover home health care visits.
2) Investigator
• The trial subject should be informed during the informed consent process about trial procedures that may take place at the subject’s home, which should be documented in the informed consent form. The trial subjects should be given the opportunity to choose a physical visit at the trial site (i.e., visit the investigator in person if needed/preferred).
• Due to subject data privacy and confidentiality, communication during Home Health Care (HHC) visits life cycle (booking, delegation of authority, conduct, and output review) should be between the investigator, subject, and the HCV/HHCP.
• The investigator is responsible for the medical care of the subjects even if the trial participant opted for HHC visits. Tasks related to medical decisions (i.e., protocol-specified medical procedures, AE/(S)AE assessment, changes in medications, etc.) should remain the responsibility of the investigator.
• The HHCP appointed for the procedure should be identified, and their tasks should be documented in the Delegation Log by the investigator, as the investigator remains ultimately responsible for the conduct and oversight of the HHC activities.
3) Registered Health Care Vendor (HCV)/HHCP
• Management of identification, qualification, and professional competency of HHCP should reside with the HCV. The HCV should also have documented procedures (SOPs) for the above HHCP identification, qualification, training, and management life cycle. Documentation and evidence such as Medical / Nursing licenses (as applicable), CVs, training certificates, and GCP certificates should be maintained.
• The HCV should ensure that appropriate guidance and training are provided to the delegated HHCP to conduct the tasks at home correctly and the training is documented. For example:
o Protocol-specific training, General Home Health Care training & data capture training. These materials should be co-developed and approved by the Sponsor.
o SOPs and training material for managing of home emergencies and life-threatening signs and symptoms should also be considered.
• Effective lines of communication between the Investigator and the HHCP who manage the subject’s HCV visit should be established in advance, there should be procedures in place to ensure the investigator is constantly kept informed in relation to trial participant safety.
• Activities conducted at the subject’s home should be adequately documented in accordance with GCP requirements. The source documentation should be part of the Investigator’s source documents and ready for monitoring, audit, and inspection in accordance with applicable regulations.
In conclusion, this guidance is intended to bring up points of consideration and improve the sponsor and investigator oversight for Home Care visits. Home Care visits can be an effective strategy to enable patients who normally are unable to participate, to be included in life-saving clinical trials by reducing the need to travel to trial sites, while increasing compliance and retention.
Collection & Delivery of Biological Samples from Trial Participants
1) Introduction Collection & delivery of biological samples from trial participants can be either planned as a standalone service for samples collected from trial participants to the site, like 24hr Urine Samples, thus reducing the trial participant’s burden. The service can be bundled with Home Care visits to maximize operational logistics, costs, and efficiencies. There could be multiple samples with different temperature requirements, so the process could entail considerable coordination and planning.
2) Risk Assessment where it is intended for the samples to be collected from the trial participant’s home, the sponsor should complete a risk assessment to determine if such an approach is appropriate. The risk assessment should, at a minimum, take into account the following aspects:
a) The feasibility and burden of the trial participant collecting and personally delivering samples to the site.
b) Sample post-collection treatment that is needed.
c) Samples stability prior, during, and/or post collection in accordance with the Lab Manual.
i) Pre-treatment of the sample collection container (e.g., chilled the blood collection tubes in ice prior to collection)
ii) Priority and urgent post-collection processing (e.g., samples to be processed within 30 minutes of collection)
iii) Specific post-collection processing (e.g., clotting time of 30-60 minutes before processing)
iv) Specific storage condition post-processing (e.g., storage at -80˚ C – dry ice) with potentially hazardous material handling
d) Requirement of temperature monitoring at specific temperature ranges for various sample types throughout the delivery journey.
e) Access to the trial participant’s data should be restricted. Information should be made available only for scheduling the collection and delivery of the samples. (Malaysia DCT Guidance Document 20)
3) Sponsor Responsibilities
a) Sponsor should consider during the planning stage of the clinical trial how appropriate collection or delivery of sample collection from the trial participants’ homes is required based on the risk assessment done above.
b) The sponsor is responsible for ensuring collecting and delivering the biological samples is performed by a qualified service provider.
c) A written contract between Sponsor and the appointed service provider must clearly establish each party's duties. Booking and collection of the biological samples shall be arranged by site personnel directly with the appointed service provider and trial participant.
4) Investigator Responsibilities
a) Investigator should provide documented instructions to the trial participant on self-collection, post-collection process, packing, storage, and completing the Laboratory Requisition Form of the sample at the trial participant’s home before the collection by the service provider. The instructions should be realistic and feasible, and the trial participant's additional burden should be documented in medical records.
b) Trial participants should be notified with their consent documented that their contact details will be used for the collection of the sample from the trial participant’s home.
c) Details regarding the use of contact information by the service provider should be outlined in the participant information sheet or informed consent form. If this was not performed during initial consent, reconsent should be obtained.
5) Service Provider Responsibilities
a) Contracted service provider is responsible for the:
i) Liaising with the site and trial participant for the collection of the sample
ii) A process of confirming the identity of the trial participant before accepting the prepacked samples
iii) It may involve the final packing of the pre-packed biological samples into the validated packaging
iv) Ensuring that the sample is delivered within the scoped turnaround time
b) The service provider is also responsible for the exportation document and application process in accordance with the standard biological exportation process but not limited to the following:
(1) Site Export License
(2) Custom Declaration
(3) Proforma Invoice
(4) Chain of Custody Form
(5) Packing List
(6) Ministry of Health MOH, BLESS Endorsement
(7) Master / House Airway Bill
c) A process whereby confirmation to the Investigator and Sponsor that the samples had been delivered and received and no temperature excursion has occurred needs to be in place.
d) All trial participants’ information is blinded except to the personnel directly involved in arranging the sample collection. The service provider should also have a data retention policy on retaining and deleting such trial participants’ information.
A well-executed DCT study may require the seamless delivery of the IP, the HHCP to conduct the visit, and orchestrate the delivery of the samples back to the site or to the central laboratory simultaneously.
7.11 Considerations/Requirements for HHC - Home Lab Collection
See Section 7.10 above.
7.12 Considerations/Requirements for Direct-to-Patient Study Product Delivery
The requirements for provisions of IP direct to the patient are described in the Clinical Research Malaysia guideline as summarized below:
Another aspect of decentralized trials involves administering Investigational Product (IP) at the trial participant’s home. At present, only IP delivery from sites to trial participants is allowed.
IP delivery from sites to trial participants is not possible for IP that is considered as a controlled substance, due to its strict control in accountability (e.g., psychotropic drugs).
The process of manufacturing, importing the IPs, and delivering the IPs directly to trial participants must comply with GxP requirements, local laws, and regulations.
Investigational Product (IP): A pharmaceutical form of an active ingredient including plant/animal-derived medicinal products or placebo being tested or used as a reference in a clinical trial, including a product with a marketing authorization when used or assembled (formulated or packaged) in a way different from the approved form, or when used for an unapproved indication (off-label use), or when used to gain further information about an approved use. For the purpose of this guidance, IP also involves comparator and Standard of Care (SOC) products.
Risk Assessment
Where it is intended for the IP to be delivered and administered at the trial participant’s home, a risk assessment should be completed and documented by the sponsor to determine if such an approach is appropriate.
The risk assessment should, at a minimum, take into account the following aspects:
• Knowledge of the IP safety profile (phase, known/possible adverse reactions, etc.), including the risk of serious adverse reactions that demand emergency treatment if, for example, allergic reactions occurs after injection. This process should be described/documented in the trial protocol.
• Develop procedures for monitoring and managing adverse events related to the self-administration of IPs at home, including documentation and reporting requirements.
• The IP route of administration and the need for healthcare professional’s assistance and subsequent observation.
• Consider the trial participant population when determining the suitability of self-administration at home, considering factors such as the trial participant's ability to self-administer the IPs and the need for careful monitoring by the investigator.
• Ensure that procedures for delivery of IPs and self-administration at home are documented, with clear instructions provided to trial participants. In addition, the IPs are labeled with instructions for use, including storage and handling instructions.
• There is a need to maintain trial participants’ anonymity and appropriately limit unnecessary outside access to information. The dispensing site and the service provider must collaborate to ensure the correct medication gets to the correct trial participant using minimal identifiers (eg: name, address, contact number) to safeguard trial participant information, in accordance with PDPA. There should be no further access to the minimal trial participant identifiers as soon as the final delivery is completed.
• Trial participant identifiers should not be made available to any parties outside its intended purposes. All efforts should be made to limit unauthorized access or disclosure of this information. The minimal identifiers of the participant (e.g., name, address, contact number) should only be shared with the service provider and not to the Sponsor.
• Access to the trial participant’s personal data should be restricted. Information should be made available only for the purpose of monitoring, auditing, and inspections if the need arises.
Expectation of implementation
Before delivering the IP to the trial participants, the below recommendations should be considered:
A. Logistics and Delivery
• Sponsor should consider during the planning stage of the clinical trial how the appropriate storage and transport conditions of the IP can be met and whether the IP is suitable for administration at home.
• Protocol should include provisions related to the adequacy of the trial participant’s home for IP storage if applicable, such as temperature control and restricted access where necessary.
• The sponsor may consider providing additional equipment necessary for the safe administration, use, and safety disposal procedure to the trial participants, in which case this should be described in the protocol or other protocol-related document (e.g., pharmacy manual), including the documentation provided to the trial participants.
• Sponsor should have a documented procedure for delivering and administering IP at the trial participant’s home.
• Investigator should provide instructions to the trial participants on using and storing the IP. The instructions should be realistic and feasible. Instructions should also be applicable for ancillary supplies that come with the IP (if applicable).
• Trial participants should be notified (documented) prior to implementing IP delivery that their contact details will be used for delivery purposes (including contact details to third parties) if the IP is to be delivered to the trial participant’s home.
• Details regarding the use of contact information by service providers should be outlined in the participant information sheet, informed consent form, or via any documentation (e.g., patient letter). Consent should be obtained from the trial participant.
• In instances whereby the trial participant (or a representative) is not available to receive the IP, the IP should be returned by the service provider to the sender (investigator’s site) in accordance with proper storage and IP handling conditions.
• There must be a written contract between the Sponsor or other third party (e.g. home health provider) and the appointed service provider that clearly establishes each party’s duties. It is recommended that the number of separate transportation steps is minimized, and principles of data privacy are adhered to. Delivery of the IP shall be arranged by site personnel directly with the appointed service provider.
• The investigator is responsible for all treatment decisions. Any decision should be documented in a source document (for example, prescription or Interactive Response Technology System) prior to dispensation and delivery of IP to the trial participant.
• IP should only be handed over to the trial participant (or a representative, if applicable) or the health care professional involved in the clinical trial. Full chain of custody documentation should be available, including IP dispensation and IP receipt.
• Storage and temperature monitoring from IP dispensation to IP receipt needs to be consistently executed and not compromise the IP, endanger the trial participant, or damage credibility in accordance with applicable regulation. Documentation (including temperature log data) related to this step should be made available.
B. Processes and Communication
• Site staff should randomize and dispense the first batch of the IP to the participant at the site. In addition to the IP labeling, clear instructions for use, including storage and handling instructions, should be provided and explained to the participants during the first dispensing.
• Delegated site staff need to check on the IP condition before releasing it to the vendor for delivery. IPs should be double-checked before they are prepared and released for delivery to trial participants. Process and documentation shall be established whereby one delegated site staff prepares the drug while another site staff counterchecks it.
• A process whereby confirmation that the correct IP has been received and no temperature excursion has occurred, needs to be in place prior to IP release to trial participant (or representative). This could be done via a phone call.
• If it is anticipated that the trial participants will prepare and administer the IP at home as outlined in the protocol, they should be instructed in advance.
• Where appropriate, clear and simple instructions are to be provided, in addition to what is already present on the IP label or package leaflet. These instructions should be adopted to the needs of the individual trial participants and documented in the source documents.
• In the event that IP is delivered directly to the trial participant's home but the IP is to be administered by healthcare personnel, clear instructions should be given to the trial participant to not administer it before the healthcare personnel's visit.
• In the event of transport temperature excursion, clear instructions should be provided to the participant not to open the IP and to quarantine it in a cool, dry place with restricted access. Trial participants should notify the site immediately for further guidance. The site should consult with the sponsor and notify the trial participant whether the trial participant could continue utilizing the IP; or wait for the delivery of the next batch.
C. Drug Accountability and Compliance
• Based on Malaysian GCP, documentation of IP accountability and trial participant’s IP compliance is the investigator’s responsibility. IP logs should be completed on a real-time basis, capturing all IP dispensation and IP returns, if applicable.
• Procedures should be in place for IP return from the trial participant’s home and destruction of the unused IPs. The procedure should also cover IP recalls during the conduct of the trial, and the steps taken to avoid that the IP remains at the trial participant’s home beyond the envisaged treatment period, in compliance with the protocol and local safety requirements. In conclusion, this guidance is intended to bring up points of consideration and improve the sponsor oversight for IP delivery directly to trial participants. If proper due diligence is undertaken, IP delivery to trial participants can be an effective tool for conducting DCT in Malaysia moving forward.
7.13 Considerations/Requirements for the Use of Telemedicine
Telemedicine is legal in Malaysia; the guidelines are set out in the Telemedicine Act 1997.
The use of telemedicine in a trial is subject to EC approval.
The Clinical Research Malaysia guideline does not refer specifically to telemedicine.
7.14 Considerations/Requirements for the Use of Wearables
The Clinical Research Malaysia guideline makes reference to the use of wearables as an accepted element of decentralized trials subject to appropriate measures for data capture and management.
According to the Malaysian Guideline for Good Clinical Practice (GCP), the data recorded during the clinical trial should be accurate, credible, reliable, and verifiable, regardless of the method/mode of data collection. The following important attributes of source data and records should be followed:
- Accurate
- Legible
- Contemporaneous
- Original
- Attributable
- Complete
- Consistent
- Enduring
- Available when needed
In decentralized clinical trials, there is a shift in the method of data collection and implementation of new technologies such as applications, wearables, and devices supporting direct data capture from the trial participants and/or their caregivers and/or service providers (e.g., home nurses). Direct data collection using electronic systems [e.g., electronic Case Report Forms (CRFs), electronic Patient Reported Outcomes (ePROs), wearables, applications, etc.] may occur, for instance, at the clinical trial site or off-site locations.
Considerations for implementation
A. Data collection – Electronic System(s)
When using electronic system(s) for direct data collection, the sponsor should ensure and implement adequate oversight and measures including, but not limited to:
• Ensure that all parties involved in the clinical trial have an overview of the data flow; a data flow diagram with additional explanations in the protocol is highly recommended.
• Ensure that the used data collection tools are created in a controlled manner, configured, and validated in accordance with their intended use. Appropriate change control and ongoing validation are needed.
• Determine the type and scope of the trial participants’ personal data to be collected and ensure adequate protection in compliance with the relevant Malaysia regulations.
• Ensure that when source data captured by a data collection tool is transferred to another location and subsequently deleted from the data collection tool, both the data and the metadata are transferred (refer to Malaysian GCP).
• Implement measures such as encryption to minimize the risk of unauthorized access, when transferring the data from a data collection tool to a server.
• Ensure access to trial data is controlled by defined user rights and methods of access for all relevant parties involved. Unauthorized access should be prevented using appropriate security measures (e.g., firewalls).
• Ensure control of and continuous and complete access by the investigator to both source data generated either on-site or off-site as well as source data reported to the sponsor (e.g., central laboratory data).
• Appropriate measures and procedures should be in place to minimize the risk of erroneous data entry for data measured and entered directly by trial participants, especially on primary, key-secondary, or safety endpoints. In summary, when electronic systems are used as a mode of data collection, the principles of data collection, handling and storage requirements as stated in the Malaysian GCP and applicable regulatory requirements must be adhered to. In addition, the data protection requirements according to the relevant Malaysia regulations, including data privacy and cybersecurity laws and regulations.
• Communication equipment, which has communication network facilities or custom requirements, which may include fixed and wireless equipment would need to be certified before it can be used.
• Certification activities for communication equipment are carried out by SIRIM QAS International Sdn BHD (SIRIM QAS), which is a registered certifying agency with the Malaysia Communications and Multimedia Commission (MCMC). For further information, refer to the FAQ on the MCMC website.
7.15 Considerations/Requirements for Remote Monitoring
The requirements for remote monitoring are described in the Clinical Research Malaysia guideline and are summarized below:
Remote monitoring here refers to the act of monitoring by sponsor personnel or representatives (e.g., clinical monitors, data management personnel, or statisticians when the activity of source data review or verification is done at a location other than the sites at which the clinical investigation is being conducted (not physically on site).
Remote monitoring processes can duplicate many of the capabilities of on-site monitoring if implemented well. The overarching goal of this section is to enhance or provide alternatives to onsite monitoring so that trial participants’ safety and clinical trial data quality are maintained through different methods of monitoring. Effective monitoring of clinical investigations by sponsors personnel or representatives is critical to the protection of human subjects and the conduct of high-quality clinical trials.
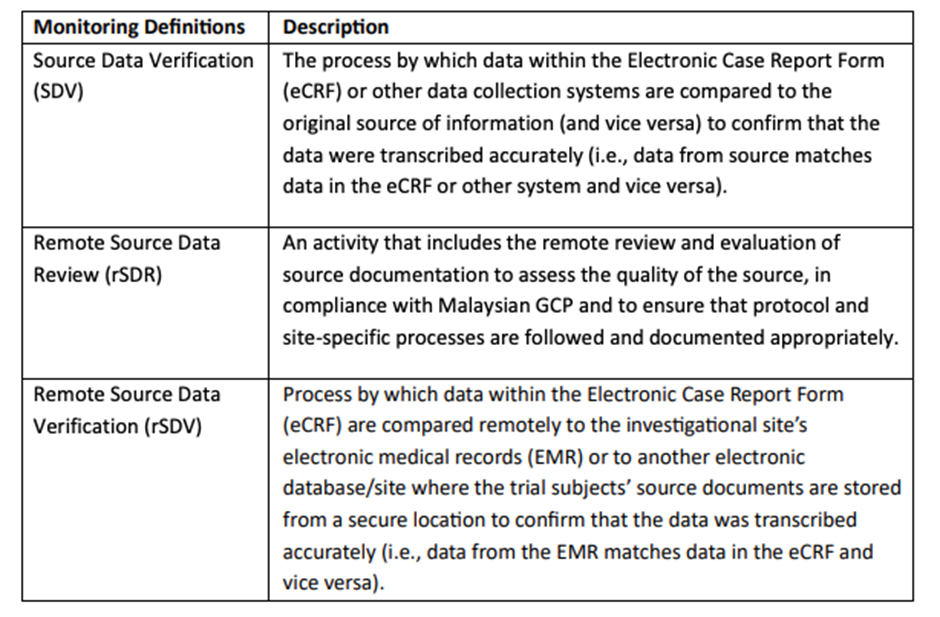
The types of monitoring activities and the extent to which remote monitoring practices can be employed depend on various factors including, but not limited to, the following:
o Limited monitoring space
o Available technological capabilities (e.g., the sponsor’s use of electronic systems; the sponsor’s access to subjects’ electronic records, if applicable)
o Additional monitoring of critical data points (e.g., the timeliness of data entry, safety events)
o Additional communication/training specific to source data entry requirements, remote subject reconsent assessments
o No access for on-site monitoring (e.g., due to pandemic/flooding)
o Policies at the respective trial sites
At present, remote monitoring can be conducted provided adequate measures are taken to ensure trial participants' confidentiality and data integrity.
Considerations for implementation
• The use of remote monitoring, including rSDV, can supplement monitoring plans to enhance data quality; real-time monitoring oversight and monitoring requirements should be documented.
• Malaysian Good Clinical Practice (GCP) states that upon request of the monitor, auditor, IRB/IEC, or regulatory authority, the investigator/institution should make available direct access for all requested trial-related records. Direct access can also refer to remote access of the data.
• The sponsor personnel or representatives are responsible for ensuring that remote monitoring including rSDV complies with Malaysian GCP and will maintain trial participant data privacy in conjunction with country's personal data and privacy act, clinical trial agreement, and hospital requirements.
• It is recommended that Informed Consent Forms (ICF) contain language that allows for remote access to medical records and corresponding privacy language clauses are present. If remote monitoring is to be conducted for a particular clinical trial, it is recommended to have it stated in the ICF.
• Establishing remote access must be in accordance with the principles of necessity and proportionality and must always be done in a way that protects the rights of the participants and does not place unnecessary burden on site staff.
• Remote monitoring should follow the principles of on-site monitoring where access to trial participants' medical records is restricted only to trial participants based on their consent. The research site should make every effort to limit the risks of any privacy or data breaches.
• The sponsor personnel or representatives should make every effort to ensure this process is performed with the highest integrity. The remote access must be traceable (i.e., audit trail of who accessed the records trial participants or equivalent documentation).
• Every effort should be made by all parties to assess the benefits and manage any risks of implementation.
• The establishment of remote access to source data should be documented. The process of remote access of source data should be documented and agreed upon by sponsor personnel or representatives, investigator, institution, and applicable departments at the site. (Malaysia DCT Guidance Document 27)
• Access must be established under secure conditions. This includes a secure connection on a machine protected from unauthorized access. The sponsor personnel or representatives should consider the quantity and types of source data that need to be accessed remotely.
• The sponsor personnel or representatives should not make unauthorized copies (e.g.: screenshots) or store personal data about the trial participants on their computers.
• The sponsor personnel or representatives’ remote access shall only be granted when necessary and be terminated immediately when the need for remote access is no longer present.
• Communication between the sponsor personnel or representatives and the study site staff is an essential component of remote monitoring. Various modes of communication could be used for this purpose (e.g., teleconferences, videoconferencing, email), provided it is a secure platform. In conclusion, remote monitoring is intended to improve the quality and efficiency of sponsor oversight of clinical trials although it is acknowledged that the process in Malaysia is not consistently established yet, hence the recommendations of this section.
For more information, please refer to the FDA Guidance for Industry Oversight of Clinical Investigations — A Risk-Based Approach to Monitoring, August 2013 Procedural or any relevant applicable regulatory requirements.
7.16 Considerations/Requirements for Digital Health Technologies (such as Platforms)
There is no specific guidance around the use of digital health technology platforms. This forms part of the review and assessment by the RA and ethics committee.
Was this article helpful?