2. General Questions
- 7 Mins to read
- DarkLight
2. General Questions
- 7 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
2.1 Name of Regulatory Authority
The Department of Health (DOH) and the Food and Drug Administration (FDA).
The FDA is an agency under the DOH that is responsible for ensuring the safety, efficacy, and quality of food, drugs, cosmetics, and medical devices.
2.2 Name of Ethics Committee
Philippine Health Research Ethics Board (PHREB) is the national policy-making body in health research ethics. An ERB/IRB is accredited based on the recommendation of the PHREB.
2.3 Clinical Trial Application Language
All documents to be submitted shall be written/officially translated into English.
2.4 Is regulatory approval required from both regulatory authorities and/or EC?
Yes; approval is required from both the regulatory body and EC.
2.5 Can regulatory authority and EC submission be done in parallel?
No; applications need to be approved by the FDA first and then forwarded to IRB/ERB. Once approved by IRB/ERB, the application is again forwarded to FDA to complete the final approval.
2.6 Requirement of any import permit/license before investigational product/study product is shipped from point of origin
As a rule, only drugs that are covered by a Certificate of Product Registration (CPR) from the FDA can be imported into and distributed in the Philippines. By way of exception, for purposes of clinical trial use, medicines not yet registered with the FDA can be imported by obtaining an import permit for investigational drug products ("Import Permit") with the FDA. In addition to unregistered drug products for clinical trials, the Import Permit allows the inclusion of ancillary supplies, such as laboratory kits, reagents, and other materials to be used for the clinical trial concerned.
Under Circular 07, the following persons may apply for the Import Permit:
- Principal investigator
- An authorized representative of the study sponsor (a registered pharmaceutical company with a permanent address in the Philippines)
- A CRO with a permanent Philippine address representing the sponsor through a letter of authorization
To obtain the Import Permit, the application must be supported by the FDA document attesting to the approval of the clinical trials to proceed based on compliance with ethical and technical requirements ascertained by the ERC.
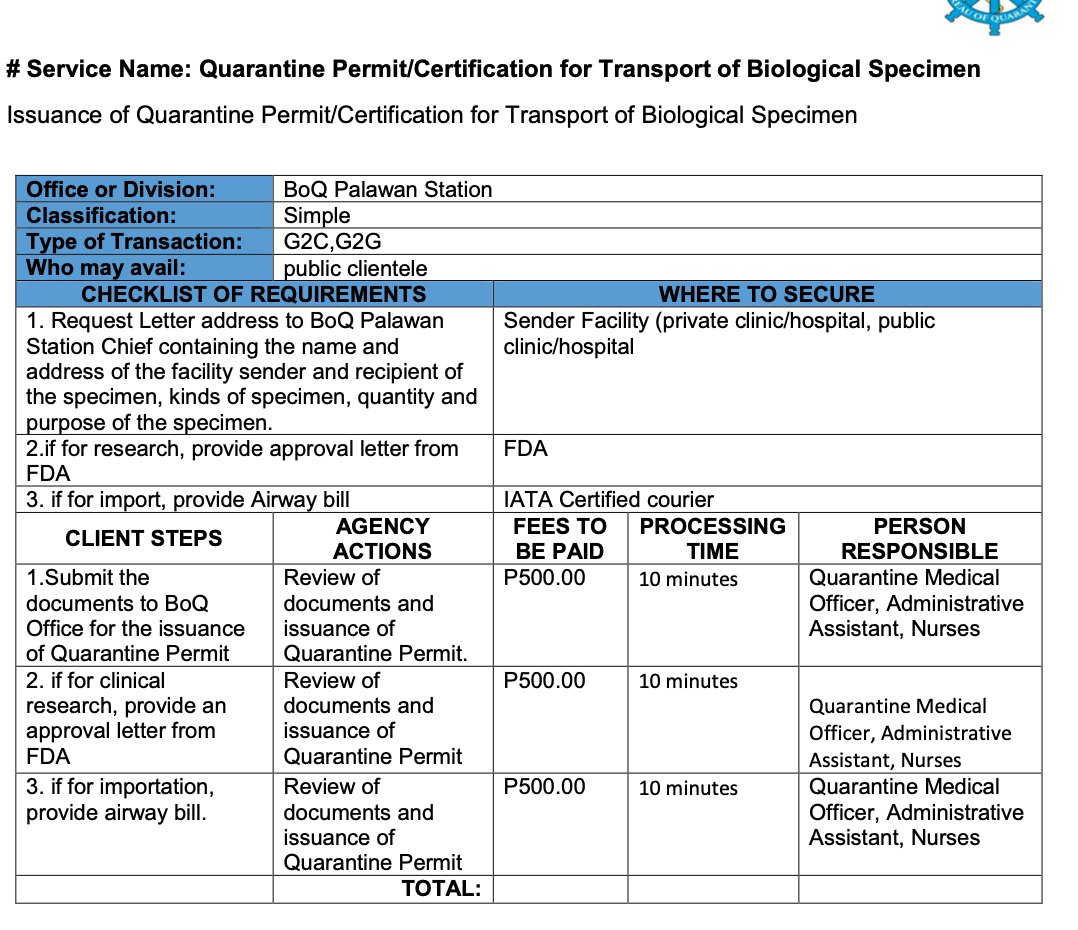
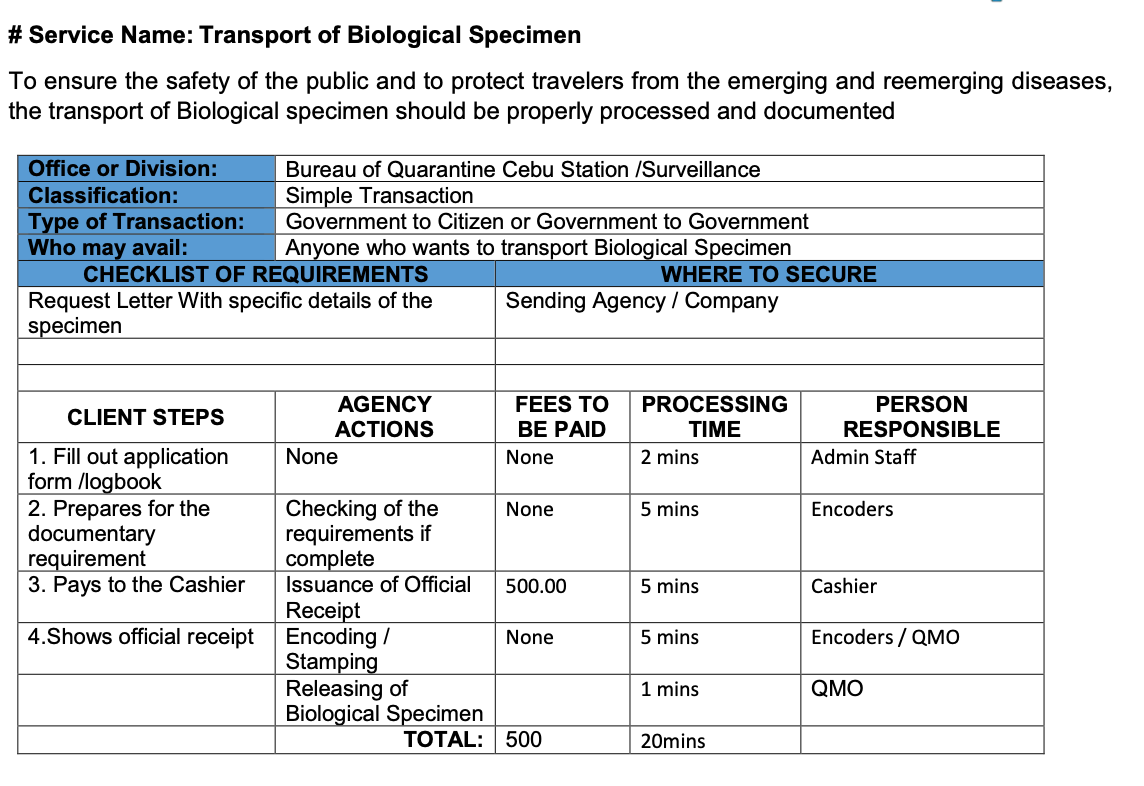
2.7 Biological Specimen Export Requirements
In the Philippines, regardless of purpose, all biological specimens must secure Quarantine Clearance before transportation.
Bureau of Quarantine - Citizen’s Charter:
INTERNATIONAL HEALTH SURVEILLANCE DIVISION COMPLEX TRANSACTION
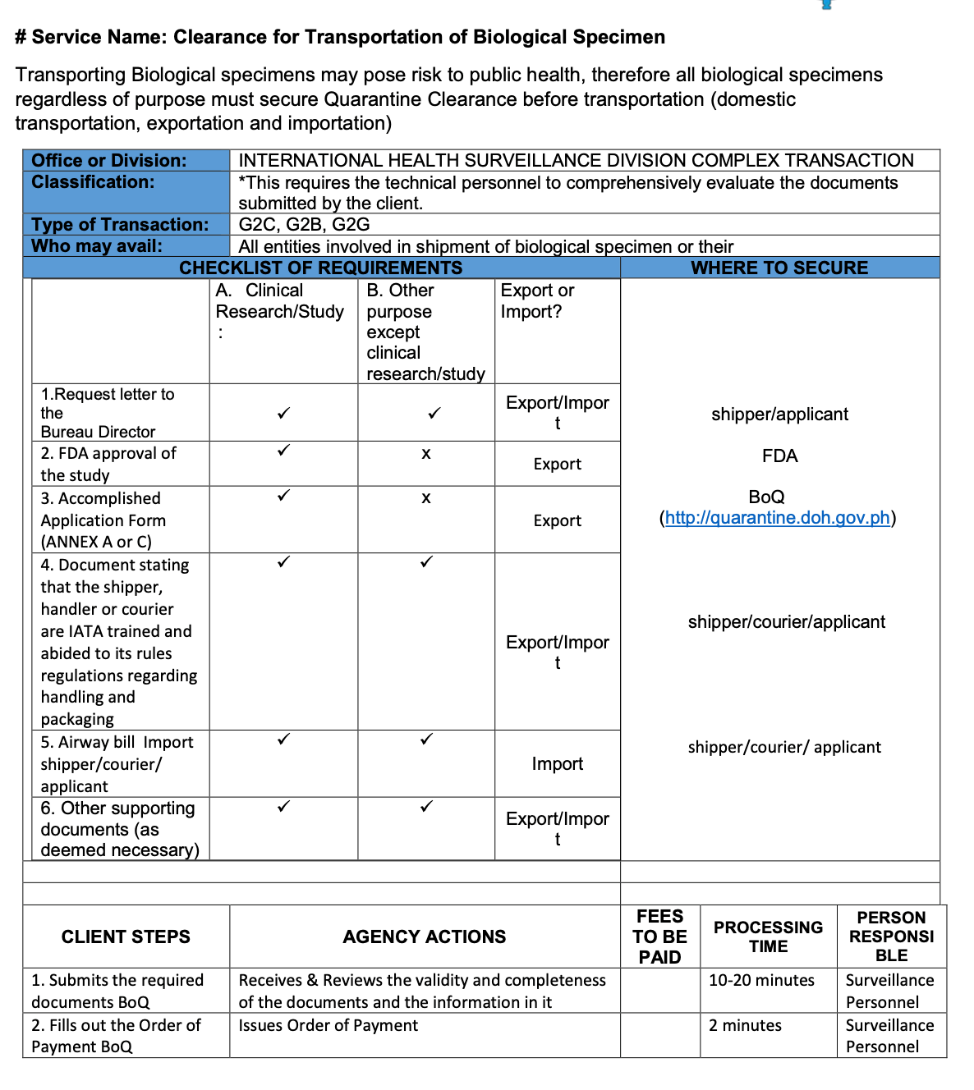
Different forms are available for each of the quarantine stations (LAOAG, PALAWAN, CEBU, and BACOLOD) as presented below:
1. LAOAG STATION


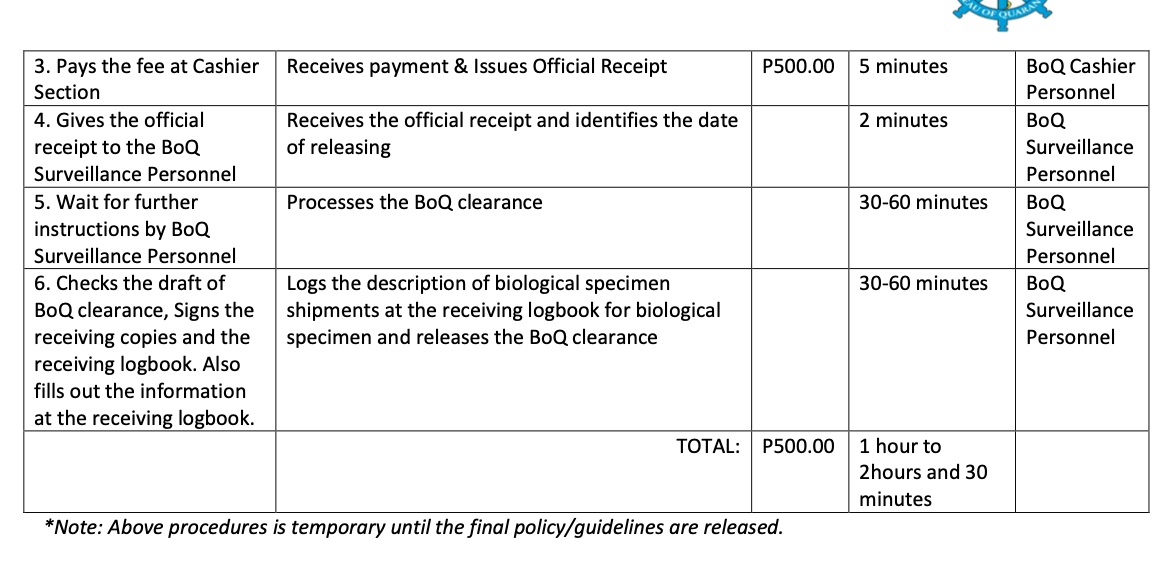
2. PALAWAN STATION
3. CEBU STATION
4. BACOLOD STATION
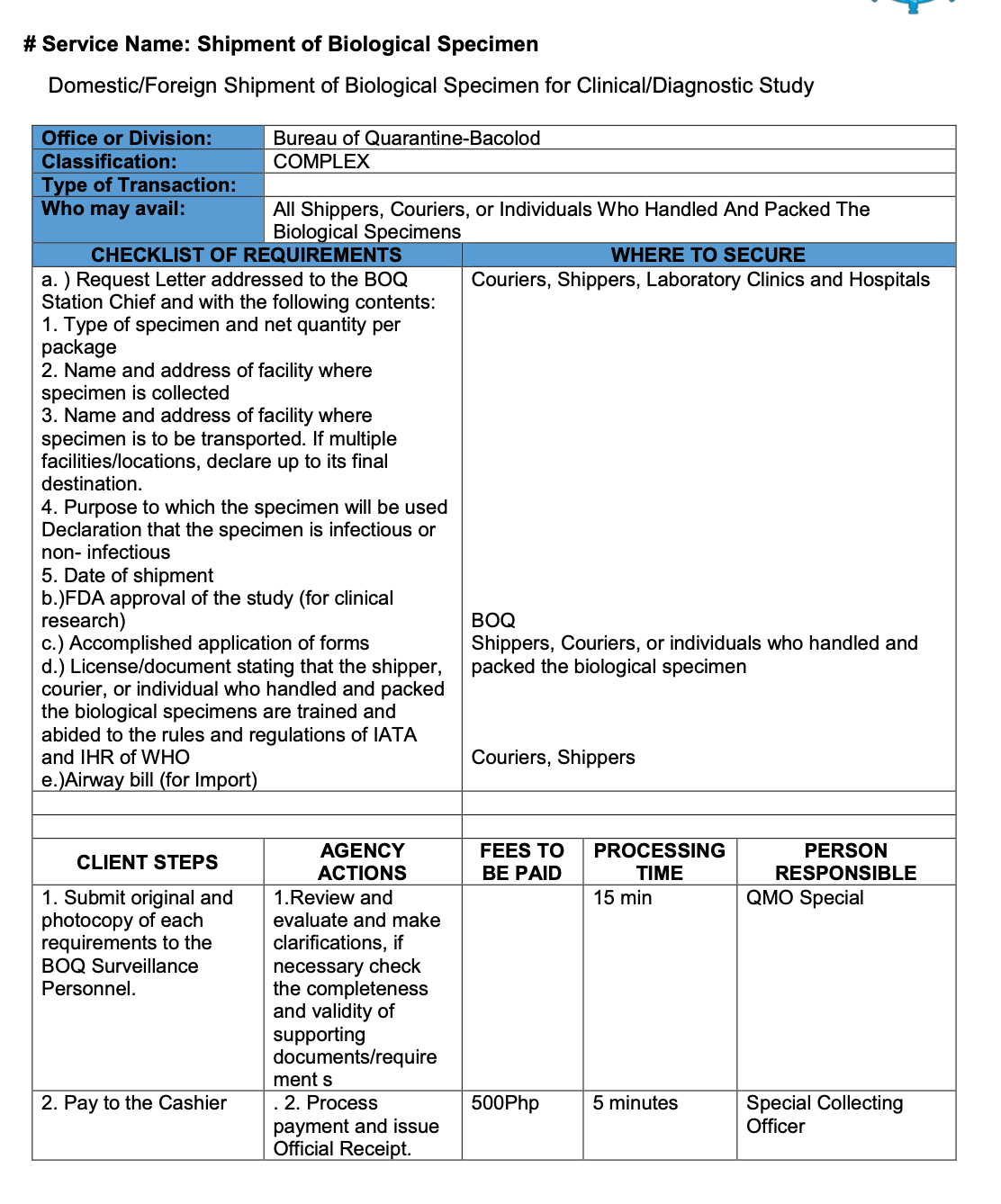
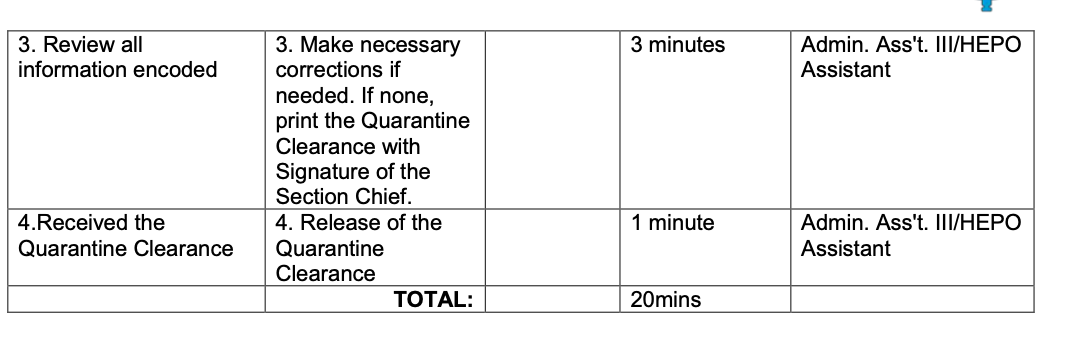
https://quarantine.doh.gov.ph/wp-content/uploads/2019/12/BOQ-Citizen-Charter-updated.pdf
2.8 Are studies of GMOs permitted (e.g., GMOs used in vaccines/vaccine manufacture and other modified products)?
Yes, GMOs are allowed subject to the regulations of the Joint Department Circular 2021-0001.
2.9 Is in-country sponsor presence/representation required?
No.
http://49.231.15.21/deptw13/upload/files/hercF256312041721022781.pdf
2.10 Is there any mandatory requirement to identify a local PI or can a PI be based in a foreign country?
The PI cannot be based in a foreign country.
http://49.231.15.21/deptw13/upload/files/hercF256312041721022781.pdf
2.11 Is there any mandatory requirement to identify a local chief or coordinating investigator?
No, only Principal Investigator.
2.12 If the applicant is CRO or a third party, does regulatory authority need any authorization or transfer of obligations from the sponsor? Does the authorization letter need to be notarized and/or apostilled?
The application may be submitted by the principal investigator/authorized representative/CRO, with a permanent Philippines address, representing the sponsor through a letter of authorization.
No regulations regarding authorization of the transfer of obligations were found. However, the applicant and CRO must have obtained a Licence to Operate (LTO) to run a clinical trial. The applicant will need to comply with the individual CRO’s processes when transferring over obligations.
2.13 Is there a requirement to register clinical trials on a local registry or database?
All clinical trials are required to be uploaded by the sponsor or the CRO (on behalf of the sponsor) in the Philippine Health Research Registry ("Registry"), administered by the Philippines Department of Science and Technology. The Registry is a publicly available database of newly approved health research, which includes clinical trials and non-clinical studies conducted in the Philippines.
2.14 What is the local requirement for clinical study documents archival; minimum of years for the archival, specific format followed (electronic/paper and/or both)?
Essential documents should be retained for at least 2 years after the last approval of a marketing application in an ICH region and until there are no pending or contemplated marketing applications in an ICH region or at least 2 years have elapsed since the formal discontinuation of clinical development of the investigational product. These documents should be retained for a longer period, however, if required by the applicable regulatory requirements or by an agreement with the sponsor. It is the responsibility of the sponsor to inform the investigator/institution as to when these documents no longer need to be retained.
2.15 Requirements around periodic safety reporting and timelines on SAEs/AEs/ADRs/SUSARs/DSURs.
The Principal Investigator must report to the ethics panel all SAEs according to the following timelines consistent with FDA Guidelines on Safety Reporting (FDA Circular 2012-007):
- Fatal or life-threatening unexpected adverse drug reactions that occurred onsite must be reported to the ethics panel promptly, no later than 7 calendar days after first knowledge by the PI that a case qualifies, followed by as complete a report as possible within 8 additional calendar days, to coincide with reporting to the FDA.
- All other serious unexpected adverse drug reactions must be reported to the ethics panel promptly, no later than 15 calendar days after first knowledge by the PI that a case meets the minimum criteria for expedited reporting, to coincide with reporting to the FDA.
- For onsite serious adverse events that are expected and non-life threatening, and all other unexpected serious adverse drug reactions that occurred off-site, a summary listing must be reported to the ethics panel attached to the submission of the progress report or final report, whichever will be submitted earlier.
Deaths MUST be reported to the ethics panel if they occur within thirty (30) days of the study intervention. Any death occurring greater than 30 days after the last dose of the investigational agent/intervention requires expedited reporting if it is possibly, probably, or definitely related to the investigational agent/intervention or if death cannot be determined based on WHO Causality Assessment definitions.
The Principal Investigator submits a copy of the SAE submission to the ethics panel coinciding with the sponsor’s timeline of the reporting requirement to the FDA.
2.16 Requirement on any periodic clinical study update, specific template, and its frequency (e.g., interim or annual progress report and final report, etc.)?
All Philippine Clinical trials are to be conducted in accordance with the latest International Council of Harmonization (ICH) and Good Clinical Practice (GCP) guidelines.
The investigator should submit written summaries of the trial status to the IRB/IEC annually or more frequently if requested by the IRB/IEC.
The investigator should promptly provide written reports to the sponsor, the IRB/ and, where applicable, the institution, on any changes significantly affecting the conduct of the trial and/or increasing the risk to subjects.
https://database.ich.org/sites/default/files/E6_R2_Addendum.pdf
2.17 Does the local regulation require notification of “serious breaches” of GCP or the trial protocol?
Yes, significant breaches of GCP should be reported to the FDA or would be subject to the following regulations relating to breaches identified during an audit.
Termination of Clinical Trial and Sanctions
For the effective implementation of Circular 2012-007, the office shall order the termination of an ongoing clinical trial without the need for a hearing should the result of random trial site inspections reveal any major violation(s), notifying only the concerned establishment of such termination. Other sanction(s) to concerned entities shall be imposed respectively under the following instances of violations:
1. The result of the random clinical trial site inspections shall have the following categories:
- No violation - no objectionable conditions or practices were found during the inspection, or the significance of the documented objectionable conditions found does not justify further FDA action (from USFDA). Compliant with GCP rules and approved protocol.
- Minor violations - regulatory violations uncovered during the inspection are few and do not seriously impact subject safety or data integrity.
- Major violations- the regulatory violation(s) uncovered is/are significant/serious and/or numerous, and the scope, severity, or pattern of violation(s) support a finding that:
- Subjects under the care of the investigator would be or have been exposed to an unreasonable and significant risk of illness or injury.
- Subjects’ rights would be or have been seriously compromised or data integrity or reliability is or has been compromised.
- Non-disclosure of conflict of interest by the investigator and other members of the trial team.
- Failure to get an informed consent is a major violation.
Any pharmaceutical product the clinical trial of which has been ordered terminated by FDA shall be a ground for the invalidation of data for drug registration purposes and, accordingly, disapproval of subsequent application for product registration pursuant to Paragraphs (1) or (6), Item B, Section 4, Article I, Book II of the Implementing Rules and Regulations of Republic Act No. 9711 because application requirements do not meet the technical requirements or appropriate standards, or such other analogous grounds or causes as determined by the FDA.
2. Disciplinary actions shall be imposed on the following after finalizing the Inspection Report by the Legal Division of the FDA.
2.18 Does RA/CA require insurance and indemnity to cover the sponsor and investigator’s potential liability?
The National Ethical Guidelines provide that a clinical trial protocol must, among others, discuss financing and insurance. There are no specific guidelines on the level of insurance required; this is subject to definition by the sponsor and CRO and approval by the ethics committee.
Was this article helpful?