3. Ethics Committee (EC)/Institutional Review Board (IRB)
- 9 Mins to read
- DarkLight
3. Ethics Committee (EC)/Institutional Review Board (IRB)
- 9 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
3.1 Provide a list of documents needed to be submitted for EC/IRB review and approval including the number of copies and/or translations as relevant.
Each EC has its own application form and process. The documents required for submission are generally as follows:
- Cover letter to the Member Secretary
- Type of review requested
- Application form for initial review
- Informed consent document (in English and the local language(s)) including translation and back translation certificates, if applicable
- Case record form/questionnaire
- Recruitment procedures (e.g., advertisement, notices) if applicable
- Patient instruction card, diary, etc., if applicable
- IB (as applicable for drugs, biological, or device trials)
- Details of funding agency/sponsor and fund allocation, if applicable
- Investigators’ Curriculum Vitaes (CVs)
- Conflict of interest statement, if applicable
- Good Clinical Practice (GCP) training certificate for investigators (preferably within the last five (5) years)
- Any other research ethics/other training evidence, if applicable as per EC standard operating procedures (SOPs)
- List of ongoing research studies undertaken by the principal investigator, if applicable
- Investigator’s undertaking statement with all participating investigator signatures
- Regulatory permissions (as applicable)
- Relevant administrative approvals (such as the Health Ministry’s Screening Committee (HMSC) approval for international trials)
- Institutional Committee for Stem Cell Research (IC-SCR) if applicable
- Memorandum of Understanding (MoU) in case of studies involving collaboration with other institutions, if applicable
- Clinical trial agreement between the sponsors, investigator, and the head of the institution(s), if applicable
- Clinical trial registration documentation (preferable)
- Insurance policy (it is preferable to have the policy as well as the insurance certificate) for study participants indicating conditions of coverage, date of commencement, and date of expiry of coverage of risk (if applicable)
- Indemnity policy, clearly indicating the conditions of coverage, commencement date, and expiry date of risk coverage (if applicable)
- Any additional document(s), as required by EC (such as other EC clearances for multicentric studies)
- Protocol
See page 34 of ICMR Ethical Guidelines 201 for a list of documents to be submitted for EC review.
https://clinregs.niaid.nih.gov/country/india#submission_content
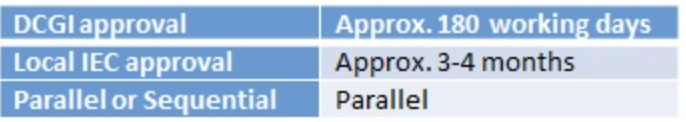
3.2 Time required for EC/IRB review and approval process and turnaround time if any query is raised during the review process.
The Independent Ethics Committee (IEC) review and approval process, which can occur at the same time as the DCGI review, generally takes 4 to 6 weeks. The study site is responsible for the initial submission of the documents and responses required by the Independent Ethics Committee (IEC). The IEC submission is made under the requirements of the local committee.
https://pharmaphorum.com/views-and-analysis/regulatory-timelines-asia-pacific/
3.3 Does EC/IRB have any fast-track or expedited review process?
Yes. Proposals that pose no more than minimal risk to patients may undergo expedited review. For example:
- Research involving non-identifiable specimens and human tissue from sources like blood banks, tissue banks, and left-over clinical samples;
- Research involving clinical documentation materials that are non-identifiable (data, documents, records);
- Modification or amendment to an approved protocol including administrative changes or correction of typographical errors and change in researcher(s);
- Revised proposals previously approved through expedited review, full review, or continuing review of approved proposals;
- Minor deviations from originally approved research causing no risk or minimal risk;
- Progress/annual reports where there is no additional risk (e.g. activity limited to data analysis). An expedited review of SAEs/unexpected AEs will be conducted by the SAE subcommittee.
- For multicentre research where a designated main EC among the participating sites has reviewed and approved the study, a local EC may conduct only an expedited review for site-specific requirements in addition to the full committee common review.
https://main.icmr.nic.in/sites/default/files/guidelines/ICMR_Ethical_Guidelines_2017.pdf
3.4 Does EC/IRB need to be registered and/or accredited/approved by RA/CA?
Yes. ECs must be registered with the Drug Controller General of India (DCGI).
3.5 How frequently do EC/IRB meet?
Each EC has its own meeting schedule, but this can vary greatly so the selection of a site and its EC is an important factor in site selection. Some ECs meet weekly whereas others only meet on an ad hoc basis or when the need arises.
3.6 Is any additional approval required apart from the EC/IRB (e.g. scientific committee, subject matter expert committee, etc.)?
No, there is no further approval required for clinical trials. However, approval is also required from the relevant committee for GMO products.
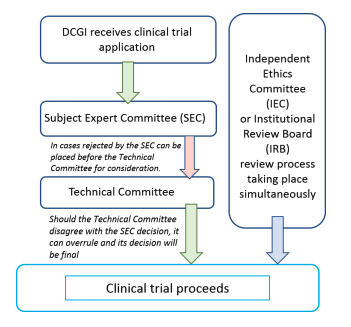
3.7 Process of EC/IRB Submission for Clinical Trial Approval
The clinical trial process in India is as follows:
3.8 What is the relevant EC/IRB fee in local currency/USD? (e.g., is the fee different for notification/approval, initial submission, amendment, study with IMPD and study without IMPD, investigator site addition, etc.)?
Ethics committee fees are set by each individual committee and therefore vary. The fee for the review of a new protocol is typically around 150,000 rupees (equivalent to USD 1800 at the time of publishing).
3.9 Does EC/IRB accept checks or can payment be made electronically? Please provide details on (1) A/C number; (2) A/C Name; (3) Sort Code; (4) Swift Code; (5) Bank address, etc. Where can remittance advice notices be sent?
These details are dependent on the local ethics committee and may vary. Therefore, there are no national regulations regarding payment which will be decided by the ethics committee.
https://clinregs.niaid.nih.gov/country/india#submission_process
3.10 Is there any guidance tool available for making electronic applications? If yes, provide the link and/or step-by-step instructions.
No, India has a decentralized ethics committee application process. Therefore, the application is dependent on the local EC institute used.
https://clinregs.niaid.nih.gov/country/india#submission_process
3.11 Does EC/IRB require any mock screens/screenshots of participant-facing material on the app? If yes, do these need to be submitted in the local language?
Yes. If electronic patient-facing materials are to be used, these should be submitted in the local language for ethics review.
3.12 Does the EC/IRB have any template or specific element requirements on ICF and/or other participant-facing materials?
There is no specific template for the ICF but it should contain the following elements:
- The study involves research and an explanation of its nature and purpose.
- The expected duration of the participant's participation.
- Any benefits reasonably expected from the research to the participant or others; if no benefit is expected, the participant should be made aware of this.
- The disclosure of specific appropriate alternative procedures or therapies available to the participant.
- The mechanism by which confidentiality of records identifying the participant will be maintained and who will have access to the participant’s medical records.
- An explanation about whom to contact for trial-related queries, participant rights, and in the event of any injury.
- The policy on compensation and/or medical treatment(s) available to the participant in the event of a trial-related injury, disability, or death.
- Note that participation is voluntary, the participant can withdraw from the study at any time, and refusal to participate will not involve any penalty or loss of benefits to which the participant is otherwise entitled.
- Any reasonably foreseeable risks or discomforts to the participant resulting from his/her participation.
- The approximate number of participants enrolled in the study.
https://clinregs.niaid.nih.gov/country/india#required_elements
3.13 Is there any guidance tool on participant compensation including clinical study-related injury per local requirements?
The sponsor (applicant), is responsible for providing compensation to research participants and/or their legal heir(s) in the event of trial-related injuries, permanent disability, or death. In the event the investigator/institution becomes the sponsor in a clinical trial, it is the host institution’s responsibility to provide compensation for research-related injury or harm as determined by the ethics committee (EC).
The sponsor (applicant) must also ensure that participants who suffer any trial-related injuries be provided with free medical treatment for such injuries as long as required per the opinion of the investigator (and the EC), or until such time it is established that the injury is not related to the clinical trial, whichever is earlier. If the sponsor or his/her representative fails to provide medical management, the Drugs Controller General of India (DCGI) must issue a written order, after a hearing, to suspend or cancel the study or restrict the sponsor including his/her representative to conduct any further clinical trial or take any other action for such period deemed appropriate for this case.
The sponsor (applicant) is responsible for compensating the research participant and/or his/her legal heir(s) if the trial-related injury, death, or permanent disability to a participant is specifically related to any of the following reasons:
- Adverse effects of an IP
- Any trial procedures involved in the study
- A violation of the approved protocol, scientific misconduct, or negligence by the sponsor, his/her representative, or the investigator
- Failure of the IP to provide the intended therapeutic effect where, the standard care, though available, was not provided to the participant per the protocol
- Not providing the required standard care, though available to the participant per the protocol in the placebo-controlled trial
- Adverse effects due to concomitant medication excluding standard care necessitated as part of the approved protocol
- Adverse effect on the child in-utero due to a parent’s participation in a trial
- Any clinical trial procedures involved in the study leading to a serious adverse effect (SAE)/serious adverse drug reaction (SADR)
- Compensation and medical management requirements are also applicable in the case of injury or death occurring during an academic trial
In the case of a trial-related injury, the sponsor (applicant) is required to provide complete medical management and compensation to the participant within 30 days of receiving an order from the DCGI. In the event of permanent injury or death, the sponsor (applicant) is required to provide compensation to the participant or to his/her legal representative(s) or guardian(s) within 30 days of receiving the DCGI’s order.
In the case of an SAE resulting in death, the DCGI must constitute an independent expert committee to review the incident and make its recommendations to the DCGI for the cause of death and to provide a quantum of compensation. The sponsor or his/her representative and the investigator must forward their reports, after due analysis, to the DCGI and the head of the institution where the trial was conducted within 14 days of the occurrence. The EC must forward its report along with its opinion on financial compensation, if any, to be paid by the sponsor or his/her representative within 30 days of receiving the investigator’s report. The DCGI, in turn, must forward the sponsor, investigator, and EC reports to the expert committee chairperson. Following its review, the expert committee must make its recommendations to the DCGI as to the cause of the SAE resulting in death and the quantum of compensation within 60 days of receiving the DCGI’s submission. The DCGI must then consider the expert committee’s recommendations and issue an order within 90 days to the sponsor or his/her representative specifying the quantum of compensation he/she is required to pay within 30 days of receiving the order.
In the case of an SAE/SADR resulting in permanent disability or any injury other than death, the sponsor or his/her representative and the investigator must forward their reports, after due analysis, to the DCGI, the EC chairperson, and the head of the institution where the trial has been conducted within 14 days of the occurrence. The EC, after due analysis, must forward its report along with its opinion on financial compensation, if any, to the DCGI within 30 days of the event occurrence. The DCGI, in turn, must determine the cause of the injury and issue an order, with the option to constitute an independent expert committee, within 60 days of receipt of the report. The DCGI must issue an order within 90 days of receiving the report indicating the quantum of compensation to be paid by the sponsor or his/her representative within 30 days of receipt of this order.
In the case of an injury not being permanent in nature, compensation should be commensurate with the participant’s loss of wages.
In the event that a sponsor or his/her representative fails to provide compensation to a research participant for trial-related injuries, or to his/her legal heir(s) in case of death, the DCGI must, after giving an opportunity to show cause why such an order should not be passed by written order, suspend or cancel the clinical trial, or restrict the sponsor or his/her representative from conducting any further clinical trials in India or taking any other action deemed fit given the circumstances.
https://main.icmr.nic.in/sites/default/files/guidelines/ICMR_Ethical_Guidelines_2017.pdf
3.14 Are there any specific local safety reporting requirements for clinical studies?
Yes. Please refer to Section 2.15 of this guidebook.
3.15 Does the EC/IRB require any periodic study reporting?
An ethics committee (EC) may periodically request study progress reports from the investigators. The length of periodic reporting differs depending on the local ethics committee.
https://cdsco.gov.in/opencms/opencms/system/modules/CDSCO.WEB/elements/download_file_division.jsp?num_id=NDI2MQ==
Was this article helpful?