6. Registration Processes
- 14 Mins to read
-
DarkLight
6. Registration Processes
- 14 Mins to read
-
DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
Food Supplements
Australia

This section provides information on any product notification or registration and manufacturing site registration processes.
6.1 Product Notification
N/A.
Products need to be entered on the relevant part of the Australian Register of Therapeutic Goods (ARTG) through a marketing approval application, not via a notification.
6.2 Product Registration
Product registration is required and is handled by the Therapeutic Goods Administration (TGA) [1]. The marketing authorization is via the approval of submission and entry of the medicine onto the Australian Register of Therapeutic Goods (ARTG). The pathway for all three types of Complementary medicine differs, as detailed below.
Listed Complementary Medicines (AUST L)
These are entered on the part of the ARTG for listed medicines, following an electronic submission which is quite abbreviated. They can only contain ingredients from a pre-approved positive list. The system automatically validates the application for quality and safety based on the formulation, use of pre-approved manufacturers, and certification by the submitter that they meet the relevant quality parameters. The efficacy is not assessed. However, the submitter attests to holding suitable evidence to support the indications which are drawn from a positive list of low-level indications. Post-market reviews of a percentage of submissions are designed to monitor industry compliance.
Listed Assessed Complementary Medicines (AUST L(A))
These are entered on the part of the ARTG for listed medicines, as described above. However, the sponsor also submits efficacy information for the assessment of the indications, which can include low-level claims (as above) and must have at least one intermediate-level claim. The draft label is reviewed by the TGA as part of this marketing authorization process. Post-market reviews of a percentage of submissions are designed to monitor industry compliance with quality and safety requirements. Since this is a newer type of application, sponsors can apply to have an existing AUST L medicine approved as an AUST L(A) by submitting the efficacy package.
Registered Complementary Medicines (AUST R)
These are entered on the part of the ARTG for registered medicines, following a full submission which is assessed for quality, safety, and efficacy. The ingredients must be drawn from the pre-approved list (as above) and/or ingredients in a schedule to the Poisons Standard (except schedule 4, 8, or 9) (refer to section “3. Compositional aspects” for full details and references). Indications may include low, intermediate, and high-level claims (refer to section “5. Claim requirements” for descriptions of each). To be clear, a registered Complementary medicine may be at this higher level due to higher-risk ingredients or higher-risk indications, or both. [2]
The marketing authorization process for newly Listed Complementary Medicine (AUST L)
Application Process
The application is submitted via the TGA electronic business services portal (TBS) [3]. In order to gain access to the portal, the sponsor requires a TBS account as described on the TGA website [4] (this page also describes the different roles that can be set up for employees and agents of the sponsor company within the business portal). There is a user guide [5] for the application process.
Before commencing the application, the user should ensure that the following are covered:
(1) All manufacturing sites have suitable GMP certification/licensing
(2) All ingredients are on the Permissible Ingredients list (refer to section “3. Compositional aspects” for details)
(3) Quality documentation is available to confirm the formulation (this does not need to be submitted)
(4) Product indications are taken from the Permissible Indications determination (refer to section “5. Claim requirements” for more details)
(5) There is a dossier of evidence to support the product indications (will not be submitted) (refer to section “5. Claim requirements” for more details)
(6) All product warnings and required advisory statements (relating to both ingredients and indications) are itemized according to RASML [6]
(7) The dosage instructions, storage details, allergens details, any messaging regarding tamper evidence seals, and the product name are clear.
Estimated Cost and Duration
Once the application is checked and submitted, it is queued until the application fee invoice is paid. Then it proceeds to the submission process and the AUST L is usually granted within two working days. The sponsor can then download the product summary and Certificate of Listing from the secure part of the TBS portal.
The current application fee is AU $893. An annual fee is also charged ($1,200). [7]
Documentation
No documentation is submitted. However, all documents described above should be collated and filed in the product regulatory file by the manufacturer. All documents should be kept up to date (change control, document updates, etc.) as the TGA may ask to see a particular product file during a site audit (GMP/manufacturing inspection).
The marketing authorization process for new Listed Assessed Complementary Medicine (AUST L(A)
Application Process
This type of application is relatively new so the application can be made by applying to 'upgrade' an AUST L to an AUST L(A) or by submitting a new AUST L(A). The instructions above for an AUST L are relevant. The application is submitted via the TGA electronic business services portal (TBS) [3] and the user guide [5] applies to the application process.
The application consists of much of the same information as for an AUST L but also requires an upload of administration information as described in [8]. This document provides links to relevant forms and guidelines.
Estimated Cost and Duration
Depending on the application category the timeline can be between 80 and 190 working days [9].
There is an application fee and an evaluation fee for these submissions [9]. Fees are below from Ref [7]. The annual charge is $1,231.
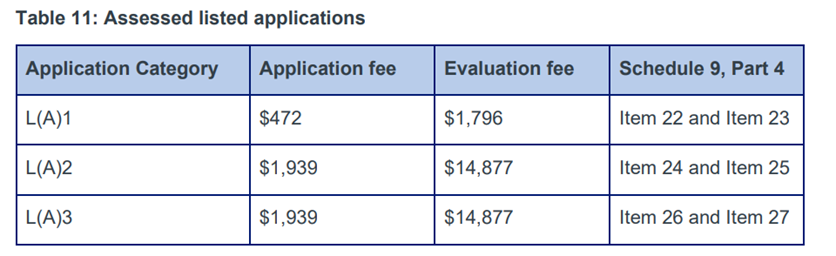
Documentation
As described under AUST L medicine and also the CTD Module 1 [8].
The marketing authorization process for new Registered Complementary Medicine (AUST R)
Application Process
Registered complementary medicine applications are categorized according to complexity. There are five levels for applications to register a complementary medicine (RCM 1 to RCM 5). Each of these levels corresponds to an application category. Applications at lower levels require less supporting information and have lower fees and reduced timeframes compared to applications at higher levels. Details on each of the levels and an overview of the process are provided in [10].
There is a guidance for the completion of CTD modules 2 to 5 [11]. Applications are submitted via TBS (refer to above under AUST L in row 15 for details). The TGA website [12] provides a matrix describing which CTD module components are required for each type of submission. Once the application is submitted and the application fee is paid the progress of the submission can be followed on TBS.
Estimated Cost and Duration
There is currently no statutory timeframe for these applications. The current application fees are in the table below. An annual fee is also charged ($1,580). [7]
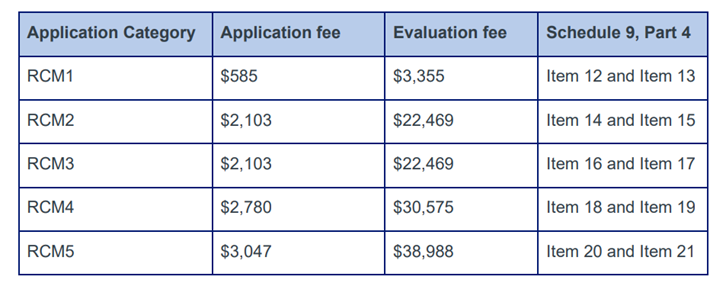 Documentation
Documentation
As described under AUST L medicine and see details in references [11].
6.3 Other Notes or Requirements
N/A.
6.4 Manufacturing Site Registration
Background Information
The term 'manufacture' has a specific meaning under Section 3 of the Act [13]: 'Manufacture, in relation to therapeutic goods that are not medical devices, means:
(a) to produce the goods; or
(b) to engage in any part of the process of producing the goods or of bringing the goods to their final state, including engaging in the processing, assembling, packaging, labeling, storage, sterilizing, testing, or releasing for the supply of the goods or of any component or ingredient of the goods as part of that process.'
Manufacturing sites within Australia require a TGA manufacturing license relevant to the medicine type, dose form, and manufacturing step. For example, a site may test non-sterile solid dose forms for chemistry and stability. Another site may manufacture and pack vitamin tablets and capsules.
Manufacturing sites outside Australia that perform manufacturing steps for Australian products on the ARTG require TGA GMP Certification. To be clear, a GMP license from an overseas regulator does not automatically qualify for GMP clearance for Australia. Overseas sites achieve TGA GMP certification via three pathways:
(1) Certification following an inspection by TGA auditors
(2) GMP clearance via the Mutual Recognition Agreement (MRA) pathway or
(3) GMP clearance via the Compliance Verification (CV) pathway.
Details on these are provided in section 6.4.1 below.
Types of products/ingredients/steps that need to be manufactured under GMP
Part 3-3 of the Act [13] covers the manufacturing of medicines. Please refer to the definition of the term, 'manufacture' under “Background Information” above for clarity on what steps this includes. Section 35 of the Act describes penalties for conducting any manufacturing step without an appropriate manufacturing license, for the goods and the steps, unless exemptions apply. This means all non-exempt manufacturing steps must be done under GMP. Exempt goods and exempt persons are permitted under Clause 34 and are described in the Regulations [14].
Schedule 7 of [14] lists therapeutic goods which are exempt and Schedule 8 lists the persons who are exempt and the therapeutics goods that apply for these:
Goods which are exempt (Sch. 7) include: (Item 2) 'ingredients, except water, used in the manufacture of therapeutic goods where the ingredients: (a) do not have a therapeutic action, or (b) are herbs, bulk hamamelis water or oils extracted from herbs, the sole therapeutic use of which is as starting materials for use by a licensed manufacturer'. This essentially excludes all excipients (except water used in manufacturing). It also excludes some active materials: bulk herbs (including farms), bulk hamamelis water, and oils manufactured from plants (including essential oils). Proprietary Ingredients (PI, refer to the section "3. Compositional aspects" for more details) which contain an active ingredient usually require manufacture under GMP since the preparation of the PI is considered to be step-in manufacture.
Note: there are other items but the above is the most relevant.
People who are exempt (Sch.8) include:
- (Items 1-4) practitioners and other professionals who manufacture (including extemporaneously) goods for patients.
- (Items 5-6) some special labeling and packaging steps.
The individual physical manufacturing sites must have their own GMP license (Australia Manufacturers) or appropriate TGA GMP clearance/certification (overseas manufacturers). This will cover the relevant manufacturing steps (can be one or many steps permitted on a license) and the types of medicines (e.g. non-sterile or sterile; dose forms; can have multiple of these on a license). A medicine must have all appropriate manufacturing steps entered on the Australian Register of Therapeutic Goods.
For example, Company A manufactures the finished medicine, and does the chemistry testing; Company B does the packaging and release for supply, and Company C does the stability and microbiology testing. For commercial reasons, there can be multiple companies approved for a single step, but the batch documents must clarify what sites were used for traceability and in case of a recall.
6.4.1 Standard/Rules for Manufacturing Site
Australia's current Therapeutic Goods (Manufacturing Principles) Determination [15] specifies that medicinal products supplied in Australia have to meet the PIC/S Guide to Good Manufacturing Practice (GMP) (01 May 2021, PE009-15) except for Annexes 4, 5, and 14 which are not adopted by Australia.
Through the operation of section 36 and other provisions within the Act [13], the PIC/S Guide to GMP has legal force in Australia. The TGA webpage provides further information, including links to the sections of the PIC/S guide.
1) Australian Sites
Manufacturing license details
A guideline [16] details the process of obtaining a license. Applications are made through the TGA business services portal (TBS). Documentation and other requirements which should be met before the application is submitted are detailed in the guideline. Once submitted and the fees paid, the application is screened. The TGA then determines if a site inspection is required or if a virtual inspection or hybrid (mix of onsite and virtual) is suitable, based on the risk profile. An inspection is scheduled, with the number of inspectors and other technical experts determined and the duration agreed upon. Once the inspection is completed, there is a closing meeting where the manufacturer is provided with an overview of any issues/deficiencies by the Lead Auditor.
The official post inspectional letter is sent (approx. four weeks after the inspection) and the manufacturer is given four weeks to respond. The TGA may ask for additional clarification, with the manufacturer given two opportunities to respond to additional TGA requests. The application is then either approved or rejected. Re-inspections are carried out on an ongoing basis based on the license expiry date. These are typically every two years but can be more or less frequent based on the compliance rating of the site.
Duration
From application to completion, the process (including the on-site inspection) can take up to twelve months. On-site inspections can typically take two to three days. The inspection close-out can take ten to thirty weeks [17]. It should be noted that the timeline targets are not being met due to the impact of Covid-19.
Estimated Costs
Application for a license AU $841. Inspection is $1,047 per hour per inspector. Holders of manufacturing licenses are required to pay an annual license charge AU$ 4,945. [7]
2) Overseas Sites
A. Certification following an inspection by TGA Auditors
Manufacturing certification details
GMP certification is usually only requested if it is not possible to obtain GMP clearance via the Mutual Recognition Agreement (MRA) or Compliance Verification (CV) pathways, for example, due to a lack of evidence. The onus is on the Australian sponsor to ensure that GMP clearance cannot be obtained via the MRA or CV pathways before applying for GMP certification.
A guideline [16] details the process of obtaining a GMP certification with an overseas inspection. Applications must be submitted by an Australian sponsor or their agent and are made through the TGA business services portal (TBS). The process is very similar to the one outlined above for Australian sites. Documentation and other requirements which should be met before the application is submitted are detailed in the guideline. Once submitted and screened, the TGA will liaise with the overseas manufacturing site to book the inspection. Upon a successful inspection, the TGA will issue a GMP certificate to the manufacturer.
Duration
From application to completion, the process (including the on-site inspection) can take up to fifteen months. The inspection close-out can take ten to thirty weeks [17]. It should be noted that the timeline targets are not being met due to the impact of Covid-19. The TGA is making use of more virtual inspections.
Estimated Costs
No application fee. Inspection is $1467 per hour per inspector plus costs and expenses (accommodation, etc.). Any invoices are sent to the Australian sponsor. [7]
B. Clearance by Mutual Recognition Agreement (MRA) Pathway
Manufacturing certification details
A guideline [18] details the process of obtaining a clearance. It should be noted that this pathway is not available for some overseas manufacturers of Complementary medicines since these products are not regulated as medicines in some jurisdictions. If this is the case, the certification process above should be followed. GMP Clearance is a non-statutory mechanism, which was introduced to reduce the regulator burden on sponsors while verifying that overseas manufacturing sites comply with the principles of GMP. GMP clearances generally expire after three years plus six months from the date of the site inspection (but may be shorter in some cases).
The MRA pathway is available if the overseas manufacturer site is located within the borders of a mutual recognition country and has been inspected by that country's regulatory authority. A list of the MRA countries is available on the TGA website [19]. This pathway can only be used if the MRA country regulator has physically inspected the manufacturing site (i.e., no desktop audits) and to a GMP standard equivalent to that used by the TGA.
The guideline [18] outlines the documentation required. In lieu of a GMP certificate, an 'Exit Notice’ or ‘certificate of GMP compliance’ (Health Canada) and a Singapore Health Sciences Authority (HSA) ‘Letter to attest GMP compliance of a manufacturer’ are acceptable. The application is made using the TGA business services portal (TBS). The guideline includes information on renewals, changes, and extensions. Renewals should be submitted no later than six months before the clearance is due to expire.
Duration
From application to completion, the target timeline is thirty working days.
Estimated Costs
- Application processing fee AU $687.
- Obtaining information from the overseas authority is $739 (if the Australian sponsor cannot obtain a copy of the overseas GMP certificate).
- Reinstatement of an expired clearance approval of $1,241. [7]
C. Clearance by Compliance Verification Pathway
Manufacturing clearance details
A guideline [18] details the process of obtaining a clearance. Please refer to the notes above under the MRA pathway regarding application processes, suitable GMP certificates, and renewals.
The CV pathway is available if the overseas manufacturer does not meet the criteria for the MRA pathway and has been inspected by a regulatory authority that has an agreement or arrangement (mutual recognition) with the TGA [19]. To be clear, this means that the regulatory authority in an approved (MRA) jurisdiction has conducted an inspection of a manufacturing site outside its borders. This pathway can only be used if the MRA regulator has physically inspected the manufacturing site (i.e., no desktop audits) and to a GMP standard equivalent to that used by the TGA.
Duration
From application to completion, the target timeline is sixty working days (non-sterile Active Pharmaceutical Ingredients (API)) and ninety working days (non-sterile finished product). [18]
Estimated Costs
- Application processing fee AU $687.
- Obtaining information (if required) from the overseas authority (US FDA only) is $739.
- Compliance verification fee $2,627.
- Reinstatement of an expired clearance approval of $1,241. [7]
6.4.2 Other Notes or Requirements for Manufacturing Site Registration
N/A.
6.5 References
1. Therapeutic Goods Administration homepage
2. TGA description of the framework
https://www.tga.gov.au/three-tiered-risk-based-framework-complementary-medicines
3. TGA Electronic business services portal (TBS)
4. TGA business services
https://www.tga.gov.au/how-we-regulate/tga-business-services-tbs
5. TGA user guide for listings on TBS
6. Required advisory statements for medicine labels (RASML). The webpage includes links the document and also guidance
7. TGA Schedule of fees and charges (updates annually, in force from 1 July)
https://www.tga.gov.au/fees-and-charges-summary-1-october-2022
8. CTD Module 1, Administrative information for assessed listed medicines
Applicable to applications received by the TGA from March 2018. Version 1.0, March 2018 (PDF supplied)
9. TGA website
10. Applications for registered complementary medicines Australian regulatory guidelines, Version 1.0, May 2020 (PDF provided)
11. CTD modules 2, 3, 4, and 5 for registered complementary medicine applications
12. Requirements for RCM applications
13. Therapeutic Goods Act 1983
https://www.legislation.gov.au/Details/C2021C00376/Download
14. Therapeutic Goods Regulations 1990
https://www.legislation.gov.au/Details/F2023C00011/Download
15. TGA webpage on manufacturing principles
16. Therapeutic Goods Administration Australian manufacturing licenses and overseas GMP certification: A step-by-step guide. V2.2, December 2021 (PDF provided)
17. Target timeframes for manufacturing inspections
https://www.tga.gov.au/target-timeframes-manufacturing-inspections
18. GMP clearance guidance. Version 18.3, July 2019 (PDF provided)
19. TGA International Agreements for GMP Clearance
Was this article helpful?