5. Claim Requirements
- 5 Mins to read
- DarkLight
5. Claim Requirements
- 5 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
Medical Devices Class I & II
Australia

This section provides information on the definition of different types of claims and any restrictions or allowance of claims as well as claim registration processes.
5.1 Overview of Claims
5.1.1 Types & Definitions of Claims
Within Australian legislation, “claims” are usually first defined by the manufacturer and are specific to a type of medical device (i.e., the Intended Use).
Medical Devices are not defined in terms of “claims and restrictions” in the same manner as medicine products.
What medical devices are “Intended For” is what they will be approved for. This is named the “Intended Purpose” and applies to all medical device classifications.
This is due to the required application of the ‘Essential Principles’ [1] to a type of Medical Device.
Application of the Essential Principles to a type of medical device, to the extent required for the Classification, is mandatory under Australian legislation before a medical device application can be made.
The Essential Principles set out the requirements relating to the safety and performance characteristics of medical devices.
There are six (6) general Essential Principles that apply to all medical devices. There are a further nine (9) Essential Principles of design and construction that apply to medical devices on a case-by-case basis. (Please refer to Appendix 1 of this guidebook).
5.1.2 List of Prohibited Claims
Important Note
This section's heading is not applicable as there are no "Prohibited Claims" for medical devices, as described below.
The Intended Purpose of a medical device is to be proven and demonstrated through evidence gained by the manufacturer and based on the evidence necessary for the class of medical device. This includes Clinical Evidence.
The specific criteria for evidence to support the Essential Principles for a certain classification of the medical device are incumbent upon the manufacturer to determine that device and more specifically the Intended Purpose and risk profile. There are only Guidelines for this - not definitive criteria.
The manufacturer is responsible for identifying relevant data and determining the extent of data needed for a complete clinical evaluation to support any Intended Purpose for the device and further demonstrate the identified safety and risk profile for that kind of medical device.
Regulators do not define what clinical data is required to support the manufacturer’s Identified Purpose of a medical device. The adopted clinical evidence ‘blueprint” for medical devices is the International Medical Device Regulators Forum.
FINAL DOCUMENT
Title: Clinical Evaluation
The defined criteria contained in that document, which manufacturers must recognize, are:
“A clinical investigation is any systematic investigation or study in or on one or more human subjects, undertaken to assess the safety, clinical performance and/or effectiveness of a medical device”.
Essential Principle 14 provides that every medical device requires clinical evidence demonstrating that the device complies with applicable Essential Principles.
Under recent changes in Australian legislation, ALL classes of medical devices now require Clinical evidence in an acceptable format for that class of medical device. This is not specified per device class as it is the manufacturer’s sole responsibility to evaluate Risk, Intended Purpose, and Patient/User profile.
The source of that data, and with reference to the:
- IMDRF MDCE WG/N56FINAL:2019 - Clinical Evaluation
- The specific claims made by the manufacturer about the safety, clinical performance, and/or effectiveness of the device
- The Classification of the medical device can be from any of the following sources -
- Existing data relevant to the clinical evaluation and held by the manufacturer or a third party having gained that from a specific clinical trial.
- Available data in the scientific literature, for the device in question or for comparable devices.
- Data generated through clinical experience including post-marketing data from existing medical devices.
- Generated from specific clinical investigations for that kind of medical device.
The manufacturer is responsible for identifying data criteria relevant to the medical device and determining the types and amount of data needed for the clinical evaluation.
For Class I-type medical devices, it is most common for a manufacturer to construct the Clinical Evaluation Report (CER) from:
- Literature sources (e.g., peer-reviewed, published medical journals and clinical papers, Cochrane database, PubMed, and similar sources).
- Post-market data from similar types of medical devices and, should that be available for the type of medical device, can also be used to construct the CER.
As a simple example, for a Class I scar reduction gel, primarily made from a silicone gel or similar combinations of Permitted Ingredients, many primary, secondary, and Meta-analysis sources exist for clinical evidence to support silicone gel as a first-line therapy for scar reduction Intended Purpose. These are assembled by or on behalf of the manufacturer, evaluated for support of the Intended Purpose, identified risk and safety profiles, and then used to satisfy the Essential principles that apply to that type of device. Additionally, that data is to be included in the technical construction file for the type of device.
For ‘Low to medium’ risk classes of medical devices including class IIa & IIb, the type and depth of clinical evidence is to be commensurate with the identified risk profile and Intended Purpose.
TGA will assess the submitted evidence and evaluate the validity of the manufacturer’s defined Intended Purpose.
If an Intended Purpose is submitted for a new medical device that does not match the corresponding evidence defined as being required for that classification of medical device, the TGA will either:
- Not commence evaluation of the application
- Request further information
- Assess though reject the application
5.2 Permitted Claims
Any Intended Purpose ‘claim’ that can be validly supported by evidence of Safety and Performance and in compliance with the Essential Principles can be submitted for assessment.
5.3 Claim Registration
Any Intended Purpose ‘claim’ that can be validly supported by evidence of Safety and Efficacy and in Compliance with the Essential Principles, i.e., Conformity Assessment [2] been applied, can be submitted as the Intended Purpose of the type of medical device as intended by the manufacturer.
This is submitted at the time of application for approval; otherwise, it is submitted later as a variation. Please refer to section 6.1 of this guidebook on Product Notification.
The process for having a medical device approved in Australia commences with the approval of the manufacturer via Conformity Assessment. Assuming that this is successfully completed, then the product application can be submitted via the online application portal.
Specific to the proposed class of medical devices, various documentation will be necessary to submit at the time of making an application.
- Class I – Australian Declaration of Conformity only
- Class I Measuring – Australian Declaration of Conformity only
- Class I Sterile - Australian Declaration of Conformity only
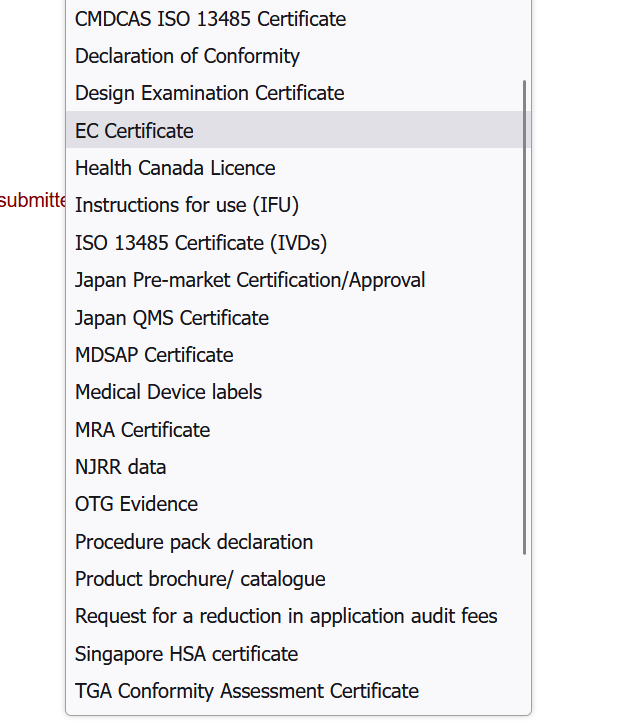
- Class IIa, IIb – any of the documents shown in the below image may be submitted as support documents
5.4 Other Notes or Requirements for Claims
During the Application audit for Class IIa and Class IIb, TGA may request additional documentation that the evaluator considers necessary for a correct assessment and approval decision on the Intended Purpose or other aspects of the application for approval. These need to be available and provided to the TGA evaluator within 20 working days usually.
5.5 References
1. The Essential Principles Checklist (Please also refer to Appendix 2 of this guidebook)
Was this article helpful?