4. Regulatory Authority (RA)/Competent Authority (CA)
- 7 Mins to read
- DarkLight
4. Regulatory Authority (RA)/Competent Authority (CA)
- 7 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
4.1 Provide a list of documents needed to be submitted for RA/CA review and approval including the number of copies and/or translations as relevant.
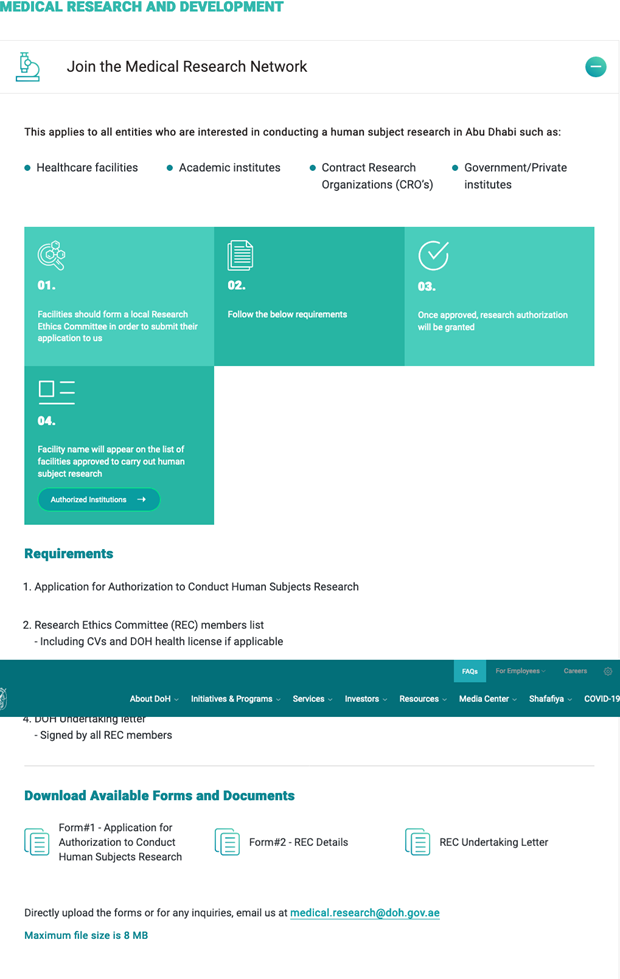
First of all, all Healthcare facilities, Academic Institutes, CROs, and Government/Private institutes wishing to conduct human research in Abu Dhabi must be registered with the DOP.
The steps to follow are indicated within the “Join the Medical Research Network” available within the DOH Medical Research and Development page.
The requirements are:
- Application for Authorization to Conduct Human Subjects Research.
- Research Ethics Committee (REC) member list, including CVs and DOH health license, if applicable.
- Research Ethics Training Course for all ethics committee members.
- DOH Undertaking letter, signed by all REC members.
All documents must be emailed to medical.research@doh.gov.ae.
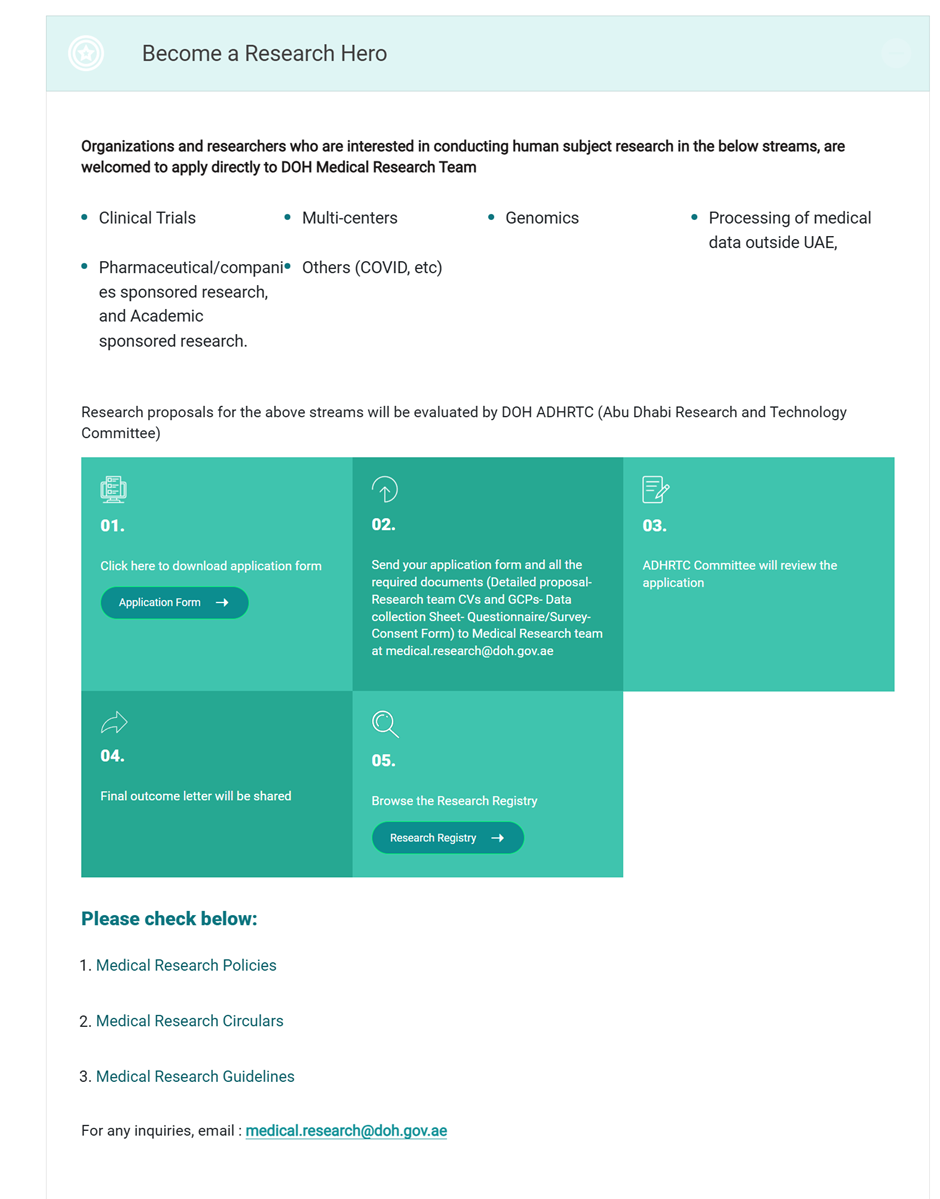
 Under the section “Becoming a Research Hero”, Organizations and researchers interested in conducting human subject research in the categories below, must submit to the DOH Medical Research Team:
Under the section “Becoming a Research Hero”, Organizations and researchers interested in conducting human subject research in the categories below, must submit to the DOH Medical Research Team:
- Clinical Trials
- Multi-centers
- Genomics
- Processing of medical data outside the UAE
- Pharmaceutical/Companies sponsored research and academic research
Research proposals for the above categories are evaluated by the DOH ADHRTC (Abu Dhabi Research and Technology Committee).
The following documents are required to be submitted for ADHRTC’s review:
Administrative Documentation:
- Ethics Committee Application (ECA): an electronic pre-defined form on an official portal that contains mandatory and optional fields and allows embedding additional files.
- Cover Letter: The applicant should submit a signed cover letter as an attachment to an ECA. The cover letter should contain the protocol number and title and a full list of all essential documents accompanying the proposed clinical trial.
- List of Regulatory Authorities and Ethics Committees apart from DOH, to which the application has been submitted, and information about their decisions.
- List of all study centers and investigators planned to participate in the Emirate of Abu Dhabi.
- Power of Attorney or Agreement authorizing the applicant of the submission on behalf of the Sponsor, in cases where the applicant is not the Sponsor of the trial.
- Statement, according to item 46 in the “Guidelines for Conducting Clinical Trials with Investigational Products and Medical Devices” document.
- Evidence of registration of the clinical trial on one of the international clinical trial data system.
- Certified copy of the Contract Research Organization (CRO).
Information about subjects
- Description of the procedures for obtaining informed consent from a legal representative, where applicable.
- Ethical grounds for enrolling participants incapable of giving informed consent, as outlined in items I, 5, 21 and 22 of the “Guidelines for Conducting Clinical Trials with Investigational Products and Medical Devices” document, where applicable.
- Any other information that will be used for subject enrolment and/or presented to patients before or during the course of a study (in English and in Arabic). Project-specific documents for the trial subjects could be any of the following:
- Patient diary
- Patient card
- Adverse Events diary
- Instructions for medication application or for handling medical device
- Scales and Questionnaires (including Quality of Life questionnaires)
- Calendar(s)
- Patient advertisement
- Additional trial information given in writing &/or multimedia technology to the subject
- Copies or pictures of any materials intended to be given to the patient
- Information for the patient/subject and Informed Consent Form (in English, Arabic, and any other language that will be used).
Documentation concerning the trial protocol
- Study Protocol and all current amendments, developed in accordance with ICH-GCP requirements and items 3 and 5 of the “Guidelines for Conducting Clinical Trials with Investigational Products and Medical Devices” document and contains as minimum:
- Clear justification of the known and potential risks and benefits, if any, to human subjects.
- Selection of Subjects – inclusion and exclusion criteria.
- Description of and justification for the selected subject population, especially in case of vulnerable subject group.
- Withdrawal of Subjects.
- Description of informed consent process in case of enrolment of subject temporary or permanent enable to be consented.
- The trial procedures to be followed, including all invasive procedures and all criteria for assessment and decisions.
- Planned monitoring and other control.
- Statistical, safety, and ethical considerations.
- Study Protocol summary in English.
- Peer review of the scientific value of the trial, where available.
- Protocol pages signed by the Sponsor and by the Investigator from each study site participating in the trial.
- Case Report form.
Documentation about the medicinal product tested
- Investigator’s brochure (issued not later than one year before application submission).
- Summary of Product Characteristics, when applicable.
- Outline/summary of all currently active clinical trials with the investigated product.
Documentation about the technical requirements and the staff
- Description of the equipment and/or the technical requirements necessary to perform the Protocol procedures.
- Certificates for external quality assessments (for the local laboratories) or Certificates for successful accreditation procedure (for the Central laboratories). Those documents are submitted for each laboratory that will be participating in the study procedures.
- CV and/or other documents confirming the qualification, experience, and training of study staff members (Investigator and Sub-Investigators) and their compliance with the requirements according to items I, 7 of the “Guidelines for Conducting Clinical Trials with Investigational Products and Medical Devices” document.
- GCP training certificates of all study staff members.
- Financial Disclosure of Principal Investigator.
- Confidentiality agreement of Principal Investigator.
- Documents, confirming the circumstances as described in items I, 8, of the “Guidelines for Conducting Clinical Trials with Investigational Products and Medical Devices” document – Accreditation of the Institution.
Data about funding and the administrative organization of trials
- Insurance covering the liability of the Sponsor and the Principal investigator(s) in case of property or non-property damages caused to the subjects related to their participation in the trial.
- Provision for compensation or a sample agreement between the Sponsor and study subjects, when such compensation is considered.
- Sample Agreement between Sponsor, Institution, and Investigator, defining terms and conditions of conducting the clinical trial.
- Written approval as outlined in items I, 8.2 of the “Guidelines for Conducting Clinical Trials with Investigational Products and Medical Devices” document – Statement by the Director of the Institution regarding permission for conducting the study (if applicable).
- Information about a clinical trial finance resource in case the Sponsor is a non-profit organization.
- Pre-site assessment report signed by the Sponsor or its representative.
- Evidence for payment of the required fee.
4.2 Time required for RA/CA review and approval process and turnaround time if any query is raised during the review process.
DOH’s “Guidelines for Conducting Clinical Trials with Investigational Products and Medical Devices” contain the following provisions regarding timelines for review:
“X. TIMELINES FOR REVIEW
- 32. The following timelines are as per international benchmarks:
- 31.4 Interventional Clinical Trial reviewed by ADHRTC: Within a period of 90 calendar days of filing an application, the REC concerned should rule, issuing an opinion, which should be sent to the applicant.
- 31.5 Non-interventional Clinical Trial reviewed by ADHRTC: Within a period of 45 calendar days of filing an application, the REC concerned should rule, issuing an opinion, which should be sent to the applicant.
- 31.6 Interventional Clinical Trial reviewed by REC/IRB: Within a period of 60 calendar days of filing an application, the REC/IRB concerned should rule, issuing an opinion, which should be sent to the applicant and ADHRTC.
- 31.7 Non-interventional Clinical Trial reviewed by REC/IRB: Within a period of 30 calendar days of filing an application, the REC/IRB concerned should rule, issuing an opinion, which should be sent to the applicant and ADHRTC.”
4.3 Does the regulation support electronic submission?
Submission of documents is via email to medical.research@doh.gov.ae.
4.4 Does the regulation require the applicant to be a Principal Investigator (PI)/Chief Investigator (CI)?
It depends on the type of trial. According to Appendix 1 - “Guideline on SOPs for Conducting Clinical Trials for Investigational Medicinal Products (CTIMPs) - of the DOH’s “Standards for Human Research”, the Sponsor is responsible for submission to the ADHRTC, but the Sponsor can delegate the responsibility to a PI or another representative.
4.5 Please describe the process of RA/CA submission for clinical trial approval.
The documents listed in Section 4.1 above must be submitted to the ADHRTC at the Department of Health-Abu Dhabi. Please follow the guidance provided in Section 4.1 above to navigate to the required information on the webpage.
The application is reviewed by the ADHRTC Committee.
A final outcome letter is sent to the applicant.
4.6 Does the RA/CA provide written acknowledgment of the submitted application? If not, describe how the application is tracked.
The application is submitted via e-mail, as described in Section 4.1 above. There is no further advice provided regarding acknowledgement of receipt of the submission, besides the fact that a final outcome letter is sent to the applicant once the application has been reviewed.
4.7 What is the relevant RA/CA fee in local currency/USD? Please provide as much information as possible (e.g. if the fee is different for notification/approval, initial submission, amendment, study with IMPD and study without IMPD, etc.)
From the research conducted, the exact application fee could not be established.
DOH’s “Guidelines for Conducting Clinical Trials with Investigational Products and Medical Devices” contain the following provisions regarding payment of fees (please note that, as a submission may be made simultaneously to the ADHRTC and REC/IRB for approval of a clinical trial application, it may be assumed that a single fee is payable to cover review by both authorities):
“28.4 The IRB/REC may collect a fee for the submission of applications requesting an opinion. The fee should be in the amount determined in the tariff.”
4.8 Does RA/CA accept checks or can payment be made electronically? Please provide details on (1) A/C number; (2) A/C Name; (3) Sort Code; (4) Swift Code; (5) Bank address, etc. Where can remittance advice notices be sent?
From the research conducted, these details could not be established.
4.9 Is there any guidance tool available for making an electronic application? If yes, provide the link and/or step-by-step instructions.
The application forms and supporting documentation are required to be uploaded via e-mail, as described in Section 4.1 above.
4.10 Does RA/CA require any screenshots/mock screens for participant-facing materials?
This is permitted, if required by the research protocol and approved by ADHRTC. Please refer to Section 7 of this guidebook.
Was this article helpful?