4. Regulatory Authority (RA)/Competent Authority (CA)
- 21 Mins to read
- DarkLight
4. Regulatory Authority (RA)/Competent Authority (CA)
- 21 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
4.1 Provide a list of documents needed to be submitted for RA/CA review and approval including the number of copies and/or translations as relevant.
The sponsor is required to submit an Investigational New Drug (IND) application to the FDA if it intends to conduct a clinical investigation with an investigational new drug. See: 21CFR312.20- Requirement for an IND.
The required documents (original + two copies, all in English, or translated into English, hard copy or electronic – refer to Section 4.3 below) are as follows (see: 21CFR312.22 – and 21CFR312.23):
- Cover sheet (Form FDA-1571). A cover sheet for the application containing the following:
- The name, address, and telephone number of the sponsor, the date of the application, and the name of the investigational new drug.
- Identification of the phase or phases of the clinical investigation to be conducted.
- A commitment not to begin clinical investigations until an IND covering the investigations is in effect.
- A commitment that an Institutional Review Board (IRB) that complies with the requirements…. will be responsible for the initial and continuing review and approval of each of the studies in the proposed clinical investigation and that the investigator will report to the IRB proposed changes in the research activity….
- A commitment to conduct the investigation in accordance with all other applicable regulatory requirements.
- The name and title of the person responsible for monitoring the conduct and progress of the clinical investigations.
- The name(s) and title(s) of the person(s) responsible…. for the review and evaluation of information relevant to the safety of the drug.
- If a sponsor has transferred any obligations for the conduct of any clinical study to a contract research organization (CRO), a statement containing the name and address of the contract research organization, identification of the clinical study, and a listing of the obligations transferred. If all obligations governing the conduct of the study have been transferred, a general statement of this transfer - in lieu of a listing of the specific obligations transferred - may be submitted.
- The signature of the sponsor or the sponsor's authorized representative. If the person signing the application does not reside or have a place of business within the United States, the IND is required to contain the name and address of and be countersigned by an attorney, agent, or other authorized official who resides or maintains a place of business within the United States.
- A table of contents.
- Introductory statement and general investigational plan.
- A brief introductory statement giving the name of the drug and all active ingredients, the drug's pharmacological class, the structural formula of the drug (if known), the formulation of the dosage form(s) to be used, the route of administration, and the broad objectives and planned duration of the proposed clinical investigation(s).
- A brief summary of previous human experience with the drug, with reference to other INDs if pertinent, and to investigational or marketing experience in other countries that may be relevant to the safety of the proposed clinical investigation(s).
- If the drug has been withdrawn from investigation or marketing in any country for any reason related to safety or effectiveness, identification of the country(ies) where the drug was withdrawn and the reasons for the withdrawal.
- A brief description of the overall plan for investigating the drug product for the following year. The plan should include the following:
- the rationale for the drug or the research study;
- the indication(s) to be studied;
- the general approach to be followed in evaluating the drug;
- the kinds of clinical trials to be conducted in the first year following the submission (if plans are not developed for the entire year, the sponsor should indicate this);
- the estimated number of patients to be given the drug in those studies; and
- any risks of particular severity or seriousness anticipated on the basis of the toxicological data in animals or prior studies in humans with the drug or related drugs.
- [Reserved]
- Investigator's brochure. If required…. a copy of the investigator's brochure, containing the following information:
- A brief description of the drug substance and the formulation, including the structural formula, if known.
- A summary of the pharmacological and toxicological effects of the drug in animals and, to the extent known, in humans.
- A summary of the pharmacokinetics and biological disposition of the drug in animals and, if known, in humans.
- A summary of information relating to safety and effectiveness in humans obtained from prior clinical studies. (Reprints of published articles on such studies may be appended when useful.)
- A description of possible risks and side effects to be anticipated on the basis of prior experience with the drug under investigation or with related drugs, and of precautions or special monitoring to be done as part of the investigational use of the drug.
- Protocols.
- A protocol for each planned study. (Protocols for studies not submitted initially in the IND should be submitted….) In general, protocols for Phase 1 studies may be less detailed and more flexible than protocols for Phase 2 and 3 studies. Phase 1 protocols should be directed primarily at providing an outline of the investigation - an estimate of the number of patients to be involved, a description of safety exclusions, and a description of the dosing plan including duration, dose, or method to be used in determining dose - and should specify in detail only those elements of the study that are critical to safety, such as necessary monitoring of vital signs and blood chemistries. Modifications of the experimental design of Phase 1 studies that do not affect critical safety assessments are required to be reported to the FDA only in the annual report.
- In Phases 2 and 3, detailed protocols describing all aspects of the study should be submitted. A protocol for a Phase 2 or 3 investigation should be designed in such a way that, if the sponsor anticipates that some deviation from the study design may become necessary as the investigation progresses, alternatives or contingencies to provide for such deviation are built into the protocols at the outset. For example, a protocol for a controlled short-term study might include a plan for an early crossover of non-responders to an alternative therapy.
- A protocol is required to contain the following, with the specific elements and details of the protocol reflecting the above distinctions depending on the phase of the study:
- A statement of the objectives and purpose of the study.
- The name and address and a statement of the qualifications (curriculum vitae or another statement of qualifications) of each investigator, and the name of each sub-investigator (e.g., research fellow, resident) working under the supervision of the investigator; the name and address of the research facilities to be used; and the name and address of each reviewing Institutional Review Board.
- The criteria for patient selection and exclusion of patients and an estimate of the number of patients to be studied.
- A description of the design of the study, including the kind of control group to be used, if any, and a description of methods to be used to minimize bias on the part of subjects, investigators, and analysts.
- The method for determining the dose(s) to be administered, the planned maximum dosage, and the duration of individual patient exposure to the drug.
- A description of the observations and measurements to be made to fulfill the objectives of the study.
- A description of clinical procedures, laboratory tests, or other measures to be taken to monitor the effects of the drug in human subjects and to minimize risk.
- Chemistry, manufacturing, and control information.
- As appropriate for the particular investigations covered by the IND, a section describing the composition, manufacture, and control of the drug substance and the drug product. Although in each phase of the investigation, sufficient information is required to be submitted to assure the proper identification, quality, purity, and strength of the investigational drug, the amount of information needed to make that assurance will vary with the phase of the investigation, the proposed duration of the investigation, the dosage form, and the amount of information otherwise available. FDA recognizes that modifications to the method of preparation of the new drug substance and dosage form and changes in the dosage form itself are likely as the investigation progresses. Therefore, the emphasis in an initial Phase 1 submission should generally be placed on the identification and control of the raw materials and the new drug substance. Final specifications for the drug substance and drug product are not expected until the end of the investigational process.
- It should be emphasized that the amount of information to be submitted depends upon the scope of the proposed clinical investigation. For example, although stability data are required in all phases of the IND to demonstrate that the new drug substance and drug product are within acceptable chemical and physical limits for the planned duration of the proposed clinical investigation if very short-term tests are proposed, the supporting stability data can be correspondingly limited.
- As drug development proceeds and as the scale or production is changed from the pilot-scale production appropriate for the limited initial clinical investigations to the larger-scale production needed for expanded clinical trials, the sponsor should submit information amendments to supplement the initial information submitted on the chemistry, manufacturing, and control processes with information appropriate to the expanded scope of the investigation.
- Reflecting the distinctions described in this [paragraph], and based on the phase(s) to be studied, the submission is required to contain the following:
- Drug substance. A description of the drug substance, including its physical, chemical, or biological characteristics; the name and address of its manufacturer; the general method of preparation of the drug substance; the acceptable limits and analytical methods used to assure the identity, strength, quality, and purity of the drug substance; and information sufficient to support the stability of the drug substance during the toxicological studies and the planned clinical studies. Reference to the current edition of the United States Pharmacopeia - National Formulary may satisfy relevant requirements in this paragraph.
- Drug product. A list of all components, which may include reasonable alternatives for inactive compounds, used in the manufacture of the investigational drug product, including both those components intended to appear in the drug product and those that may not appear but are used in the manufacturing process, and, where applicable, the quantitative composition of the investigational drug product, including any reasonable variations that may be expected during the investigational stage; the name and address of the drug product manufacturer; a brief general description of the manufacturing and packaging procedure as appropriate for the product; the acceptable limits and analytical methods used to assure the identity, strength, quality, and purity of the drug product; and information sufficient to assure the product's stability during the planned clinical studies. Reference to the current edition of the United States Pharmacopeia - National Formulary may satisfy certain requirements in this paragraph.
- A brief general description of the composition, manufacture, and control of any placebo used in a controlled clinical trial.
- Labeling. A copy of all labels and labeling is to be provided to each investigator.
- Environmental analysis requirements. A claim for categorical exclusion or an environmental assessment.
- Pharmacology and toxicology information. Adequate information about pharmacological and toxicological studies of the drug involving laboratory animals or in vitro, on the basis of which the sponsor has concluded that it is reasonably safe to conduct the proposed clinical investigations. The kind, duration, and scope of animal and other tests required vary with the duration and nature of the proposed clinical investigations. Guidance documents are available from FDA that describe ways in which these requirements may be met. Such information is required to include the identification and qualifications of the individuals who evaluated the results of such studies and concluded that it is reasonably safe to begin the proposed investigations and a statement of where the investigations were conducted and where the records are available for inspection. As drug development proceeds, the sponsor is required to submit informational amendments, as appropriate, with additional information pertinent to safety.
- Pharmacology and drug disposition. A section describing the pharmacological effects and mechanism(s) of action of the drug in animals, and information on the absorption, distribution, metabolism, and excretion of the drug, if known.
- Toxicology.
- An integrated summary of the toxicological effects of the drug in animals and in vitro. Depending on the nature of the drug and the phase of the investigation, the description is to include the results of acute, subacute, and chronic toxicity tests; tests of the drug's effects on reproduction and the developing fetus; any special toxicity test related to the drug's particular mode of administration or conditions of use (e.g., inhalation, dermal, or ocular toxicology); and any in vitro studies intended to evaluate drug toxicity.
- For each toxicology study that is intended primarily to support the safety of the proposed clinical investigation, a full tabulation of data is suitable for detailed review.
- For each nonclinical laboratory study subject to the good laboratory practice regulations under part 58, a statement that the study was conducted in compliance with the good laboratory practice regulations in part 58, or, if the study was not conducted in compliance with those regulations, a brief statement of the reason for the noncompliance.
- Previous human experience with the investigational drug. A summary of previous human experience known to the applicant, if any, with the investigational drug. The information is required to include the following:
- If the investigational drug has been investigated or marketed previously, either in the United States or other countries, detailed information about such experience that is relevant to the safety of the proposed investigation or to the investigation's rationale. If the drug has been the subject of controlled trials, detailed information on such trials that is relevant to an assessment of the drug's effectiveness for the proposed investigational use(s) should also be provided. Any published material that is relevant to the safety of the proposed investigation or to an assessment of the drug's effectiveness for its proposed investigational use should be provided in full. Published material that is less directly relevant may be supplied by a bibliography.
- If the drug is a combination of drugs previously investigated or marketed, the information required under…. of this section should be provided for each active drug component. However, if any component in such combination is subject to an approved marketing application or is otherwise lawfully marketed in the United States, the sponsor is not required to submit published material concerning that active drug component unless such material relates directly to the proposed investigational use (including publications relevant to component-component interaction).
- If the drug has been marketed outside the United States, a list of the countries in which the drug has been marketed and a list of the countries in which the drug has been withdrawn from marketing for reasons potentially related to safety or effectiveness.
- Additional information. In certain applications, as described below, information on special topics may be needed. Such information shall be submitted in this section as follows:
- Drug dependence and abuse potential. If the drug is a psychotropic substance or otherwise has abuse potential, a section describing relevant clinical studies and experience and studies in test animals.
- Radioactive drugs. If the drug is a radioactive drug, sufficient data from animal or human studies to allow a reasonable calculation of radiation-absorbed dose to the whole body and critical organs upon administration to a human subject. Phase 1 studies of radioactive drugs must include studies that will obtain sufficient data for dosimetry calculations.
- Pediatric studies. Plans for assessing pediatric safety and effectiveness.
- Other information. A brief statement of any other information that would aid the evaluation of the proposed clinical investigations with respect to their safety or their design and potential as controlled clinical trials to support the marketing of the drug.
- Relevant information. If requested by the FDA, any other relevant information needed for the review of the application.
- Information previously submitted. The sponsor ordinarily is not required to resubmit information previously submitted but may incorporate the information by reference. A reference to information submitted previously must identify the file by name, reference number, volume, and page number where the information can be found. A reference to information submitted to the agency by a person other than the sponsor is required to contain a written statement that authorizes the reference and that is signed by the person who submitted the information.
- Material in a foreign language. The sponsor shall submit an accurate and complete English translation of each part of the IND that is not in English. The sponsor shall also submit a copy of each original literature publication for which an English translation is submitted.
- Number of copies. The sponsor shall submit an original and two copies of all submissions to the IND file, including the original submission and all amendments and reports.
- Numbering of IND submissions. Each submission relating to an IND is required to be numbered serially using a single, three-digit serial number. The initial IND is required to be numbered 000; each subsequent submission (e.g., amendment, report, or correspondence) is required to be numbered chronologically in sequence.
- Identification of exception from informed consent. If the investigation involves an exception from informed consent…., the sponsor shall prominently identify on the cover sheet that the investigation is subject to the requirements in [this chapter].
4.2 Time required for RA/CA review and approval process and turnaround time if any query is raised during the review process.
In broad terms, an investigational product may be used in a clinical investigation if the following two conditions are met (21CFR312.40 - General Requirements for use of an investigational new drug in a clinical investigation):
“(b) An IND goes into effect:
- Thirty days after the FDA receives the IND unless the FDA notifies the sponsor that the investigations described in the IND are subject to a clinical hold; or
- On earlier notification by the FDA that the clinical investigations in the IND may begin. FDA will notify the sponsor in writing of the date it receives the IND; and
- Each participating investigator conducts his or her investigation in compliance with the requirements of this 21CFR312 [and 21CFR50 and 21CFR56].
(c) The sponsor may ship an investigational new drug to investigators named in the IND:
- Thirty days after the FDA receives the IND; or
- On earlier FDA authorization to ship the drug.
(d) An investigator may not administer an investigational new drug to human subject until the IND goes into effect under paragraph (b) of this section.”
The FDA may order the sponsor to delay a clinical trial or to suspend an ongoing investigation – a clinical hold 21CFR312.42 - Clinical holds and request for modification).
4.3 Does the regulation support electronic submission?
Yes. Sponsors of commercial INDs and all subsequent amendments are required to submit information electronically in the electronic Common Technical Document (eCTD) format. For electronic submission in eCTD, see Information on Regulatory Submissions in Electronic Format for Biologic Products. IND sponsors should request a submission tracking number (STN) from CBER prior to an eCTD submission. Detailed procedures are outlined in SOPP 8117: Issuing Tracking Numbers in Advance of Electronic Submissions in eCTD Format.
While non-commercial/research IND sponsors are exempt from the requirement for electronic submissions, they are encouraged to submit electronically using the eCTD format. For specific instructions on how to submit eCTD exempt non-commercial/research INDs to CBER, please refer to SOPP 8110: Submission of Regulatory Applications- Exempt from eCTD Requirements. General questions regarding the submission of an IND to CBER may be directed to industry.biologics@fda.gov.
See FDA’s guidance “Providing Regulatory Submissions in Electronic Format — Certain Human Pharmaceutical Product Applications and Related Submissions Using the eCTD Specifications” Guidance for Industry (February 2020). Commercial INDs are covered by this requirement – see the FDA’s Electronic Common Technical Document (eCTD) page. See also the FDA’s note “FDA Electronics Submissions Gateway (ESG)”, the ESG being the central transmission point for sending information electronically to FDA.
4.4 Does the regulation require the applicant to be a Principal Investigator (PI)/Chief Investigator (CI)?
No, the sponsor is required to submit an Investigational New Drug application to the FDA if it intends to conduct a clinical investigation with an investigational new drug. See: 21CFR312.20 - Requirement for an IND.
4.5 Please describe the process of RA/CA submission for clinical trial approval.
In broad terms, an investigational product may be used in a clinical investigation if the following two conditions are met (21CFR312.40– General requirements for use of an investigational new drug in a clinical investigation):
(b) An IND goes into effect:
- Thirty days after the FDA receives the Investigational New Drug (IND) unless the FDA notifies the sponsor that the investigations described in the IND are subject to a clinical hold; or
- On earlier notification by the FDA that the clinical investigations in the IND may begin. FDA will notify the sponsor in writing of the date it receives the IND; and
- Each participating investigator conducts his or her investigation in compliance with the requirements of this 21CFR312 [and 21CFR50 and 21CFR56].
Please see the following flowchart taken from the FDA’s Investigational New Drug Applications Prepared and Submitted by Sponsor-Investigators - Guidance for Industry (May 2015) -DRAFT-
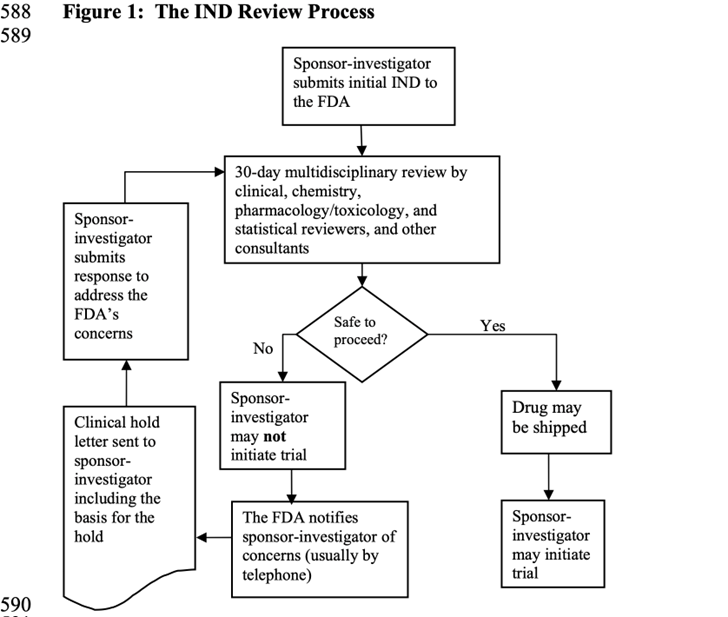
4.6 Does the RA/CA provide written acknowledgment of the submitted application? If not, describe how the application is tracked.
Yes, the FDA provides written acknowledgment.
The FDA’s IND Application Procedures: Overview states that “[u]pon receipt of an IND application, FDA will notify the sponsor of the date it receives the application through an IND acknowledgment letter.”
The FDA’s Information for Sponsor-Investigators Submitting Investigational New Drug Applications (INDs) provides that: “[u]pon receipt of the IND by FDA, an IND number will be assigned, and the application will be forwarded to the appropriate reviewing division. The reviewing division will send a letter to the Sponsor-Investigator providing notification of the IND number assigned, the date of receipt of the original application, the address where future submissions to the IND should be sent, and the name and telephone number of the FDA person to whom questions about the application should be directed. Studies shall not be initiated until 30 days after the date of receipt of the IND by FDA unless you receive earlier notification by FDA that studies may begin.”
4.7 What is the relevant RA/CA fee in local currency/USD? Please provide as much information as possible (e.g. if the fee is different for notification/approval, initial submission, amendment, study with IMPD and study without IMPD, etc.)
The FDA does not levy a fee on the submission of an Investigational New Drug (IND).
4.8 Does RA/CA accept checks or can payment be made electronically? Please provide details on (1) A/C number; (2) A/C Name; (3) Sort Code; (4) Swift Code; (5) Bank address, etc. Where can remittance advice notices be sent?
The FDA does not levy any submission fee for INDs and hence this section is not applicable for INDs submitted in USA.
4.9 Is there any guidance tool available for making an electronic application? If yes, provide the link and/or step-by-step instructions.
The FDA has published the “Electronic Common Technical Document (eCTD)”, with a link also to “Providing Regulatory Submissions in Electronic Format — Certain Human Pharmaceutical Product Applications and Related Submissions Using the eCTD Specifications. Guidance for Industry” (February 2020).
4.10 Does RA/CA require any screenshots/mock screens for participant-facing materials?
It is unusual for ICFs to be submitted as part of an IND. However, it could still be the case (see below), in those cases when a eConsent is intended to be used, the FDA could request screenshots of the digital information.
In the FDA’s final guidance on “Informed Consent” released in August 2023, it indicates the following:
“The IND regulations (21 CFR part 312) do not specifically require submission to FDA of the consent form with the IND. However, if FDA determines that review of the consent form is necessary to make the determination of whether the clinical investigation may safely proceed, the Agency will request that the sponsor submit the consent form for review under 21 CFR 312.23(a)(11).
As a general matter, FDA will review the informed consent form for treatment INDs and treatment protocols (21 CFR part 312, subpart I) and INDs for studies conducted under the Exception From Informed Consent Requirements for Emergency Research (21 CFR 50.24) for consistency with 21 CFR 50.25 (see 21 CFR 50.24(a)(6)).
For other clinical investigations of drugs, FDA often considers the following factors in determining whether to require submission and review of the consent form:
- Nonclinical studies submitted in support of the first administration of a drug in humans have identified an unusual toxicity.
- Unusual known toxicity is associated with the investigational drug, the drug class to which the drug belongs, or with a different drug with characteristics similar to those of the study drug.
- The study subject population is particularly vulnerable.
- The study design is unusual for the therapeutic class.
- The clinical investigation is a postmarketing safety clinical trial, required under section 505(o) of the FD&C Act to assess a serious risk.
- The clinical investigation has significant potential for serious risk to human subjects.
- The clinical investigation involves asking subjects to forgo or delay effective treatment that is known to decrease long-term mortality or irreversible morbidity.
- FDA has other confidential or proprietary information not available to an IRB that affects the assessment of whether the informed consent form adequately addresses risks.
After reviewing the consent materials, if the FDA review divisions have specific concerns about the adequacy or compliance of the consent materials with 21 CFR part 50, details about these concerns normally will be conveyed to the sponsor in writing. In rare circumstances, FDA may find a consent form to be misleading, inaccurate, or incomplete in a way that makes informed consent inadequate and noncompliant with 21 CFR part 50 to the extent that subjects would be exposed to an unreasonable and significant risk of illness or injury. In these cases, FDA may require that specific revisions be made to address the concern(s) before the clinical investigation can proceed (21 CFR 312.42).”
Was this article helpful?