4. Regulatory Authority (RA)/Competent Authority (CA)
- 10 Mins to read
- DarkLight
4. Regulatory Authority (RA)/Competent Authority (CA)
- 10 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
4.1 Provide a list of documents needed to be submitted for RA/CA review and approval including the number of copies and/or translations as relevant.
Whether the clinical trial involves the use of pharmaceutical, biological, and radiopharmaceutical drugs or medical devices, the following section provides the requirements for a Clinical Trial Application (CTA) or an Investigational Testing Authorization (ITA).
For the use of pharmaceutical, biological, and radiopharmaceutical drugs, Health Canada requires the sponsor to apply for clinical trial authorization by submitting a clinical trial application (CTA) to Health Canada. The CTA should be organized into three modules:
- Module 1 - Administrative and clinical information about the proposed trial
- Module 2 - Quality (Chemistry and Manufacturing) summaries about the drug product(s) to be used in the proposed trial
- Module 3 - Additional supporting quality information
The clinical trial application form and the following information and documents must be submitted:
Module 1 Administrative and Product Information
- 1.0 Correspondence
- 1.0.1 Cover letter
- 1.0.5 Meeting Information
- 1.1 Table of Contents
- 1.2 Administrative Information
- 1.2.1 Application Forms
- 1.2.3 Certification and Attestation Forms
- 1.2.5 Compliance and Site Information
- 1.2.5.1 Clinical Trial Site Information Form
- 1.2.6 Authorization for Sharing Information
- 1.2.7 International Information
- 1.2.9 Other Administrative Information
- 1.3 Product Information
- 1.3.4 Investigator's Brochure
- 1.4 Health Canada Summaries
- 1.4.1 Protocol Safety and Efficacy Assessment Template - Clinical Trial Application (PSEAT-CTA)
- 1.7 Clinical Trial Information
- 1.7.1 Protocol
- 1.7.2 Informed Consent Forms
- 1.7.3 Canadian Research Ethics Board (REB) Refusals
- 1.7.4 Information on Prior-related Applications
Module 2 Common Technical Document Summaries
- 2.1 Table of Contents
- 2.3 Quality Overall Summary (QOS)
Module 3 Quality (if submitted)
- 3.1 Table of Contents of Module 3
- 3.2 Body of Data
- 3.3 Literature References
As for medical devices, in order to conduct investigational testing in human subjects, and prior to study initiation, manufacturers and importers of Medical Devices are required to submit Investigational Testing Authorization (ITA) to Health Canada in order to sell or import a medical device.
Health Canada will issue a “letter of authorization” for investigational testing medical devices (where applicable and according to their specific class - for a Class I device, there is no requirement to apply for authorization to conduct investigational testing) if the application meets the requirements stated in Part 3 of the regulations (SOR/98-282) although the ethics committee approval may not be available at the time the ITA application review has been processed and should be submitted to HC as soon as it becomes available. https://laws-lois.justice.gc.ca/eng/regulations/sor-98-282/page-10.html#h-1021976
As per HC requirements, a “non-eCTD electronic-only” format for ITA must be filed and sent by email to devicelicensing-homologationinstruments@hc-sc.gc.ca as follow (https://www.canada.ca/en/health-canada/services/drugs-health-products/medical-devices/application-information/guidance-documents/investigational-testing-authorizations-guidance/guidance-document.html#app-4)
- 01 - Cover Letter & Executive Summary
- 02 - Table of Contents
- 03 - Application Form
- 04 - Pre-submission Correspondence
- 05 - Introduction
- 05.01 Manufacturer or Importer Identification
- 05.02 Device Identification
- 05.03 Device Description
- 05.04 Design Philosophy
- 05.05 Indications For Use, Intended Use, Contraindications
- 05.06 Device Labelling
- 05.07 Market History
- 06 - Risk Assessment and Risk Reduction Measures
- 06.01 Risk Assessment
- 06.02 Previous Studies
- 06.03 Alternate Treatments or Testing Options
- 06.04 Precautions
- 07 - Institutional Information
- 07.01 Investigator Information
- 07.02 Curriculum Vitae
- 07.03 Investigator Agreements
- 07.04 Name and Address of Institutions
- 07.05 Research Ethics Board Approval
- 08 - Study Documents
- 08.01 Protocol
- 08.02 Investigator’s Brochure
- 08.03 Informed Consent Form
- 09 - Appendices
Some sections are not applicable (to class II application). Empty folders mut be deleted before filing to HC. An appendix can be provided for any other supporting documentation that does not fall into one of the pre-defined folders (https://www.canada.ca/en/health-canada/services/drugs-health-products/medical-devices/application-information/guidance-documents/investigational-testing-authorizations-guidance/guidance-document.html).
4.2 Time required for RA/CA review, approval process, and turnaround time if any query is raised during the review process.
In accordance with Part C (Division 5) of the Food and Drug Regulations, an authorized CT is one that has been filed with Health Canada and has not received an objection within 30 days (although guidance specifies 30 days, as per experience, it could take 6 weeks from the moment HC acknowledges the receipt of a complete application, and they may take as long as 6 weeks to respond). Consideration of 4-6 weeks will be a more realistic approach.
Upon receipt of a CT application, the relevant Health Canada Directorate screens the application package for completeness. If deficiencies are found, the Directorate sends the sponsor a Request for Clarification or a Screening Rejection Letter. The sponsor has 2 days (may vary on a case-by-case basis and could take as long as 15 days) to provide the requested information. If the Directorate finds the application complete, an acknowledgment letter is issued to indicate the 30-day default review period commenced on the date of receipt.
Per the G-CanadaCTApps, once a clinical trial is authorized, the sponsor is allowed to sell or import a drug for use in a trial, if a CTA has been filed with Health Canada and has not received an objection within 30 days.
"Health Canada commits to a 30-day default, review period for authorization from the date a complete application is received. Health Canada has an administrative 7-day review target for comparative bioavailability trials, and some Phase I trials in healthy human volunteers". Refer to https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/applications-submissions/guidance-documents/clinical-trials/frequently-asked-questions-process.html On August 13, 2020, the Minister of Health approved an order to temporarily extend the default period to review clinical trial applications and amendments by 15 days https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/applications-submissions/guidance-documents/clinical-trials/covid-19-extension-notice.html.
Canada’s processing time for clinical trial applications is internationally competitive, as evidenced by this comparative graph (data published in 2014):
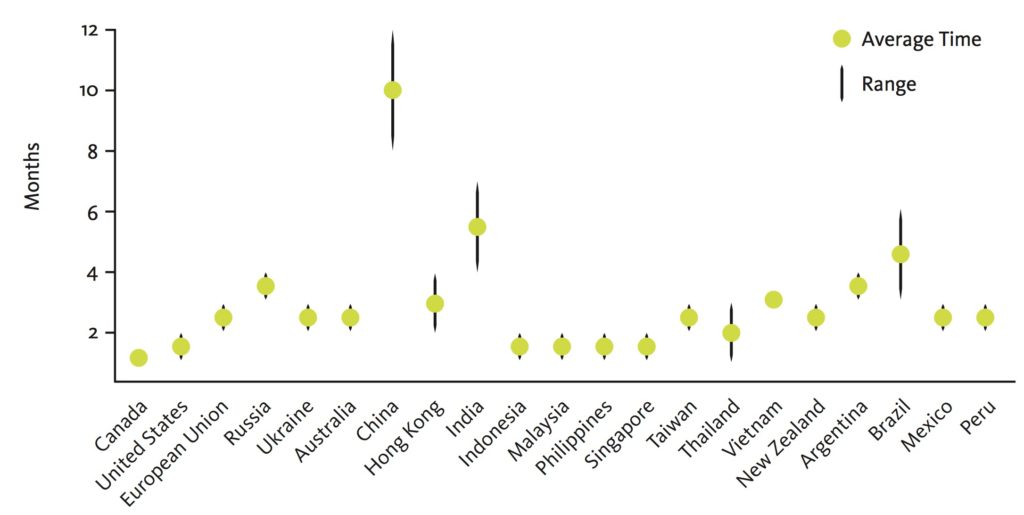
https://www.cctam.ca/resources-support/regulatory-process-in-canada/
4.3 Does the regulation support electronic submission?
Yes.
As of 1 June 2016, Health Canada no longer accepts paper copies of CT applications. Any paper CT application received will be shredded or returned at the sponsor’s expense. Each of the CT modules must be submitted in electronic format using a folder structure that easily delineates each module and sub-folders within each module (see section 4.1 above). Health Canada recommends that sponsors submit the CT application in the “Common Technical Document” format. For a complete review of the format and submission requirements, please review the Guidance document: preparation of regulatory activities in the non-eCTD format published by Health Canada, available at:
Filing an ITA application for medical devices must be also submitted electronically as specified in section 4.1 above.
4.4 Does the regulation require the applicant to be Principal Investigator (PI)/Chief Investigator (CI)? Are there any exceptions allowed?
No.
The sponsor is required to submit the clinical trial application (CTA) or the Investigational Testing Authorization (ITA). The sponsor is permitted to transfer any or all of its trial-related duties and functions to a contract research organization (CRO) and/or institutional site(s). However, the ultimate responsibility for the trial data’s quality and integrity always resides with the sponsor. Any trial-related responsibilities transferred to a CRO should be specified in a written agreement. A sponsor may be domestic or foreign. A foreign sponsor is required to have a senior medical or scientific officer who is residing in Canada who will represent the sponsor, and sign and date the application and the clinical trial attestation form.
Studies can be initiated by the PI (or QI); these studies are called Investigator Initiated Studies (IIS) or Investigator Sponsored studies (ISS). It is still the responsibility of the sponsor (of the device or the drug, etc.) to file an application to HC.
4.5 Please describe the process of RA/CA submission for clinical trial approval
A sponsor is required to obtain clinical trial authorization from Health Canada prior to initiating the trial. The sponsor must file a CT application / Investigational Testing Authorization (ITA) to the appropriate Directorate within Health Canada. In addition, the sponsor may submit a CT application for clinical trial authorization to Health Canada in parallel with its submission to an REB for a favorable ethical opinion.
Health Canada will not authorize the sponsor to begin the clinical trial until he/she submits an REB (provided in the required Clinical Trial Site Information (CTSI) form (CAN-6)) for each participating trial site. The revised CTSI form improves efficiencies and supports the submission of CTAs using the electronic Common Technical Document (eCTD) format. Instructions on filling out and submitting the CTSI are available at https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/applications-submissions/forms/instructions-clinical-trial-information-form.html.
For authorization to conduct investigational testing on human subjects in Canada, only manufacturers and importers can apply for an ITA. In either case, a senior official of the manufacturer of the unlicensed device must complete and sign the application. This is also applicable when applications are sponsored by an investigator or a third party. In such a situation, regulatory correspondence and clinical oversight can be delegated to the clinical investigator.
Regulatory Submission
CT applications should be sent directly to the appropriate Health Products and Food Branch Directorate for review—the Pharmaceutical Drugs Directorate (PDD) for pharmaceutical drugs or the Biologic and Radiopharmaceutical Drugs Directorate (BRDD) for biological drugs and radiopharmaceuticals. The outer label should be clearly identified with "Clinical Trial Application". For ITAs, please refer to section 4.1 above.
Sponsors may request a pre-submission/application meeting with the appropriate Directorate within HPFB if they have any questions or concerns prior to filing a CT application. The submission can be in French or English.
An application by a sponsor for authorization to sell or import a drug for the purposes of a clinical trial must be submitted to Health Canada, signed and dated by the sponsor’s senior medical or scientific officer in Canada and senior executive officer. The sponsor’s clinical trial attestation must be submitted with the application.
Health Canada accepts clinical trial submissions/applications in the eCTD and non-eCTD electronic-only formats. Once a submission is filed in eCTD format, all additional information and subsequent regulatory activities for the same dossier (protocol) must be filed in eCTD format, and sponsors must not revert to non-eCTD electronic-only format.
CT applications in eCTD format are available upon request via email to no-reply.ereview.non-reponse@hc-sc.gc.ca; the text 'Request for Clinical Trial Applications in eCTD Format' should be in the subject line of the email.
Health Canada’s guidance documents: “Preparation of Regulatory Activities in eCTD Format” and “Common Electronic Submissions Gateway (CESG) Health Canada Reference Guide” are available upon request via email to no-reply.ereview.non-reponse@hc-sc.gc.ca; the text ‘Request for eCTandD Guidance Document’ should be in the subject line of the email.
Applicants must request a dossier ID from Health Canada for eCTD dossiers. The dossier ID request forms for drug and biological product clinical trials are available via https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/applications-submissions/guidance-documents/filing-submissions-electronically.html. A request for a dossier ID should be sent a maximum of eight (8) weeks prior to filing a clinical trial application in the eCTD format. If the applicant is not sure what their dossier ID is they can contact Health Canada at devicelicensing-homologationinstruments@hc-sc.gc.ca. For eCTD format, prior to filing a CT application via the CESG, each company must file a sample transaction to Health Canada in accordance with the applicable guidance documents. Additional questions regarding the CESG and eCTD submissions may be directed to hc.ereview@hc-sc.gc.ca.
For non-eCTD electronic submissions, sponsors must submit CT applications on electronic media—a disc or drive. Health Canada recommends PDF and/or MS Word (where required) for the CT application. Finally, the sponsor must include a cover letter in both electronic and paper format.
4.6 Does RA/CA provide written acknowledgment of the submitted application? If not, describe how the application is tracked.
Yes.
If the Directorate finds a CT application complete, an acknowledgment letter is issued to indicate the 30-day default review period commenced on the date of receipt.
Once an ITA application is received by HC, it undergoes a screening phase for administrative and scientific content. If the information is complete and complies with sections 81 and 86 of the regulations, the review process could be initiated. The review period for a new ITA application (or request for a revised ITA, other than for combination products) is 30 calendar days from the time the screening is initiated. On the other hand, if a regulatory deficiency is identified at screening, a screening deficiency letter that details the deficiencies is issued. Missing information needs to be provided in usually 15 calendar days during the screening phase or the application would be considered withdrawn.
4.7 What is the relevant RA/CA fee in local currency/USD? Please provide as much information as possible (e.g., if the fee is different for notification/approval, initial submission, amendment, study with IMPD and study without IMPD, etc.)
There are no fees to submit a clinical trial application in Canada.
4.8 Does RA/CA accept checks or payment can be made electronically? Please provide details on (1) A/C number; (2) A/C Name; (3) Sort Code; (4) Swift Code; (5) Bank address, etc. Where can remittance advice notices be sent?
N/A. Details are provided in section 4.7 above.
4.9 Is there any guidance tool available for making electronic applications? If yes, provide the link and/or step-by-step instructions.
Yes.
As per Health Canada requirements and depending on the regulatory activity type, applicants are required to file submissions electronically to HC in either Electronic Common Technical Document (eCTD) format or non-eCTD format.
Follow the link that provides detailed information on each format along with information related to filing submissions electronically.
Health Canada, Updated - Guidance Document: Preparation of Regulatory Activities in the "Non-eCTD Electronic-Only" Format, (Ottawa, Health Canada, 2016), available at: https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/applications-submissions/guidance-documents/filing-submissions-electronically.html
4.10 Does RA/CA require any screenshots/mock screens for participant-facing materials?
Details are provided in section 4.1 above along with the discussion of Remote Monitoring in section 7.
Was this article helpful?