4. Regulatory Authority (RA)/Competent Authority (CA)
- 12 Mins to read
- DarkLight
4. Regulatory Authority (RA)/Competent Authority (CA)
- 12 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
4.1 Provide a list of documents needed to be submitted for RA/CA review and approval including the number of copies and/or translations as relevant.
As outlined in the Malaysian Guideline for Application of CTIL and CTX, the general requirements for regulatory submission are as follows:
1. Table of Contents A content page should be included in each CTIL/CTX application dossier. A template table of content can be found in Appendix A. |
2. Cover Letter The applicant shall submit a signed cover letter with the application. Its subject line should contain the full NMRR Registration Number and the protocol number with the title of the trial. In the cover letter, the applicant should draw attention to the peculiarities of the trial, if any. |
3. CTIL/CTX Application Form The applicant shall submit a complete application form with NMRR Registration Number. The application form shall be signed and dated by the applicant and stamped with the company's stamp. Application forms for CTIL (current version PPPK/SPKPK/F01) and CTX (current version PPPK/SPKPK/F02) can be downloaded from the NPRA website. Only one applicant and one local contact person from the same organization, if any, can be named under Part 2 of the application form. All communication will be sent to the named applicant and the second contact person. For CTIL/CTX applications involving FIH clinical trials, applicants are required to provide additional information as listed under Appendix D of the application form. |
4. Receipt for the processing fee, if applicable. Every application for CTIL shall be accompanied by a processing fee. The CTIL application processing fee is RM 500.00 per product. CTIL application without the correct processing fee will not be processed. The processing fee shall be paid in the form of a credit card/bank draft/money order/postal order payable to 'Biro Pengawalan Farmaseutikal Kebangsaan'. Alternatively, the payment can be made using a credit card at the Finance, Account & Revenue Section. Note: Foreign currencies are not acceptable. The processing fee is not refundable. Application for CTX is free of charge. |
5. A copy of the Company Registration Certificate, if applicable The company must be registered with Suruhanjaya Syarikat Malaysia. The applicant (if said company is not the sponsor) should be authorized in writing by the sponsor to be the holder of the CTIL/CTX. Please refer to 4.4.6 for the Letter of Authorization. A copy of the Company Registration Certificate is NOT required for an investigator-initiated trial. |
6. A copy of the applicant’s Poison Licence Types A for a pharmacist in the private sector or ARC for a public pharmacist, whichever is applicable. |
7. Letter of Authorization, if applicable
|
8. A copy of the opinion(s) of the EC which is/are registered with DCA Applications for CTIL/CTX and EC can be submitted in parallel. The favorable opinion/approval letter of EC (with attendance list) should be sent to the DCA as soon as possible when available. Following the directive issued by the DPS on Keperluan Mendaftar Jawatankuasa Etika dengan Pihak Berkuasa Kawalan Dadah, NPRA will only accept favorable opinion/approval issued by EC that is registered with the DCA. The applicant is advised to refer to the NPRA website for the current list of EC that is registered with DCA. For an application involving an FIH clinical trial, a positive opinion from EC shall be submitted to NPRA before the application can be tabled in the DCA meeting. |
9. Clinical Trial Protocol The final version of a clinical trial protocol must be submitted. The version submitted should be the version that has been submitted to EC. The clinical trial protocol shall be in the format provided by Section 6 of the Malaysian Guideline for GCP and include the definition of the end of the trial. For BE study, the formula used with detailed stepwise calculation is required to justify the sample size needed. If a two-stage design is adopted in the study, a decision tree or diagram, which depicts the methodology must be stated in the study protocol. |
10. Declaration by investigator/PI An original copy of the declaration by the investigator/ PI of each trial site should be provided. The format for the document can be found in Appendix C. The investigator protocol signature page will NOT be accepted. |
11. GCP certificate and CV for investigator/PI of each trial site It is expected that the investigator/PI will be qualified by education, approved training in GCP, and experience to assume responsibility for the proper conduct of the trial. The GCP certificate and CV for the investigator/PI of each trial site should be provided. The GCP course should be recognized/approved by NCCR, Ministry of Health Malaysia. The requirement is in accordance with the current version of the Malaysian Guideline for GCP. In the case of BE study, a GCP certificate, CV, and Declaration by the investigator for the clinical site investigator should also be provided if the investigator at the clinical site is not the PI. |
12. Informed consent form (initial version only for one of the trial sites) The ICF provided can be in either English or Bahasa Melayu. The initial version of ICF must be provided during submission. |
13. Pharmaceutical data for all products that require CTIL/CTX Quality data should be submitted in a logical structure, such as the headings of the following appendices. The following appendices outline the pharmaceutical data format for different types of IP:
Stability data It is the responsibility of the applicant and sponsor to ensure that the product used is stable for the duration of the clinical trial. A minimum of one (1) batch of stability studies under accelerated and real-time conditions for a minimum of 3 months should be provided. Stability studies should be conducted in compliance with ASEAN/ICH stability guidelines. For the First-in-Human Clinical Trial, a minimum of one (1) batch of stability studies under accelerated and real-time conditions for a minimum of 1 month should be provided. Stability data of the IP after reconstitution or dilution, if applicable, should be submitted to support the in-use period of the reconstituted or diluted IP (ICH Q1A (R2)). BE study For BE study, the test product should usually originate from a batch of at least 1/10 of the production scale or 100,000 units, whichever is higher, unless otherwise justified. Appendix D3, Section 4.S Drug Substance shall be provided only for BE study involving chemical entity that has not been registered in Malaysia. DCA-registered IP (Relabeling/Secondary Packaging Modification) In case the IP used is a DCA-registered product, the applicant is not required to submit pharmaceutical data for the IP. However, a declaration letter from the sponsor to confirm that the quality of IP is the same as the DCA-registered product should be provided. |
14. Label all products that require CTIL/CTX The applicant must ensure labels of products for clinical trials meet the labeling requirements, according to Appendix E. The particulars on the outer packaging of the investigational product, or where there is no outer packaging, on the immediate packaging, shall appear in Bahasa Melayu or English. |
15. Good Manufacturing Practice (GMP) Requirement Investigational products are required to be produced in accordance with the PICS Annex 13. A current copy of the Certificate of GMP Compliance for the manufacturer of the drug product and/or final/batch releaser only should be submitted. The name and address of the manufacturer should be identical between the application form and the GMP certificate provided. Any discrepancy in the information shall be justified. The certificate must be valid at the time of submission. Local manufacturer in Malaysia Manufacturer outside Malaysia
Note: Other proof of GMP compliance documentation can be considered based on a risk-based approach for the following conditions:
|
16. Investigator’s Brochure For content and format of the IB, reference is made to section 7 of the current version of the Malaysian Guideline for GCP. The unavailability of IB is generally acceptable for most of the BE study. However, IB shall be provided for a BE study involving a chemical entity that is not registered in Malaysia. Generally, toxicity studies are expected to be performed in compliance with Good Laboratory Practice (GLP). |
17. Overall Risk and Benefit Assessment This section should provide a brief integrated summary that critically analyzes the non-clinical and clinical data in relation to the potential risks and benefits of the proposed trial unless this information is already provided in the protocol. In the latter case, the applicant should cross-refer to the relevant section in the protocol. The text should identify any studies that were terminated prematurely and discuss the reasons. The assessment is not mandatory for a BE study. |
18. A copy of scientific advice from other regulatory agencies, if available. |
19. Evidence of Phase 1 Unit Accreditation by NPRA The Phase 1 Unit shall be on the NPRA Phase I Unit Accreditation Programme. Evidence of this listing shall be submitted for FIH clinical trial. |
20. Proof of Insurance Cover Proof of insurance shall be submitted for the FIH clinical trial for compensation for any damage suffered by the subject resulting from participation in an FIH clinical trial. |
21. Declaration by Sponsor for CTIL/CTX Application Involving FIH Clinical Trial Declaration by the sponsor shall be submitted in original copy for FIH clinical trial. A format for Declaration by Sponsor can be found in Appendix F. |
22. Electronic Format One electronic copy of all documents shall be submitted in a CD-ROM. Please refer to section 4.6.3 for the preparation of the CD-ROM document. |
23. Other or additional documents Any other trial-related documents that could be relevant for the review of the clinical trial application by DCA may be submitted, e.g. published clinical data, if applicable. |
4.2 Time required for RA/CA review and approval process and turnaround time if any query is raised during the review process.
Under normal circumstances, all CTIL/CTX applications will be assessed within the following timelines:
- 45 working days for phase I trial, including FIH clinical trials, clinical trials involving biological/biotechnological, cell therapy products, gene therapy products as well as herbal/natural products with therapeutic claims.
- 30 working days for all products except those products mentioned above.
For CTX applications, Day 0 is the day of receipt of a complete CTX application dossier. For CTIL applications, Day 0 is the day a complete CTIL application dossier AND the official receipt of payment is received.
During the evaluation phase, the evaluator may have a query raised related to the application. The clock will stop on the day the query is e-mailed to the applicant. The applicant is expected to respond to the query within 30 working days. CTIL/CTX application will be rejected if NPRA does not receive adequate response/reply for the queries or information requested by the evaluator.
The timeline for FIH clinical trials of 45 working days only accounts for CTIL/CTX evaluation by NPRA; it does not include the review time taken by the external Panel of Experts.
Fast-track reviews can be considered for the application of new IP used for treatment/prevention in pandemic/epidemic situations in the interest of public health except for FIH clinical trials.
Fast-track CTIL/CTX application will be assessed within the following timeline:
- 22 working days for phase I trial, the clinical trial involves biological/biotechnological, cell therapy products, and gene therapy products as well as herbal/natural products with therapeutic claims.
- 14 working days for all products except those products mentioned above.
4.3 Does the regulation support electronic submission?
The application to the MREC (Medical Research and Ethics Committee) is an online submission using the National Medical Research Register (NMRR) website.
4.4 Does the regulation require the applicant to be a Principal Investigator (PI)/Chief Investigator (CI)?
An application can be made by either of the following:
- The principal investigator
- The sponsor, that is, the person or entity responsible for the initiation, management, and/or financing of the trial. The sponsor must be a locally incorporated pharmaceutical company with a permanent address in Malaysia. Where the sponsor is not a local entity, it may assign its duties and functions to a Contract Research Organization (CRO) incorporated in Malaysia. The CRO may then apply on behalf of the sponsor, conditional upon the submission of a letter of authorization from the sponsor.
4.5 Please describe the process of RA/CA submission for clinical trial approval.
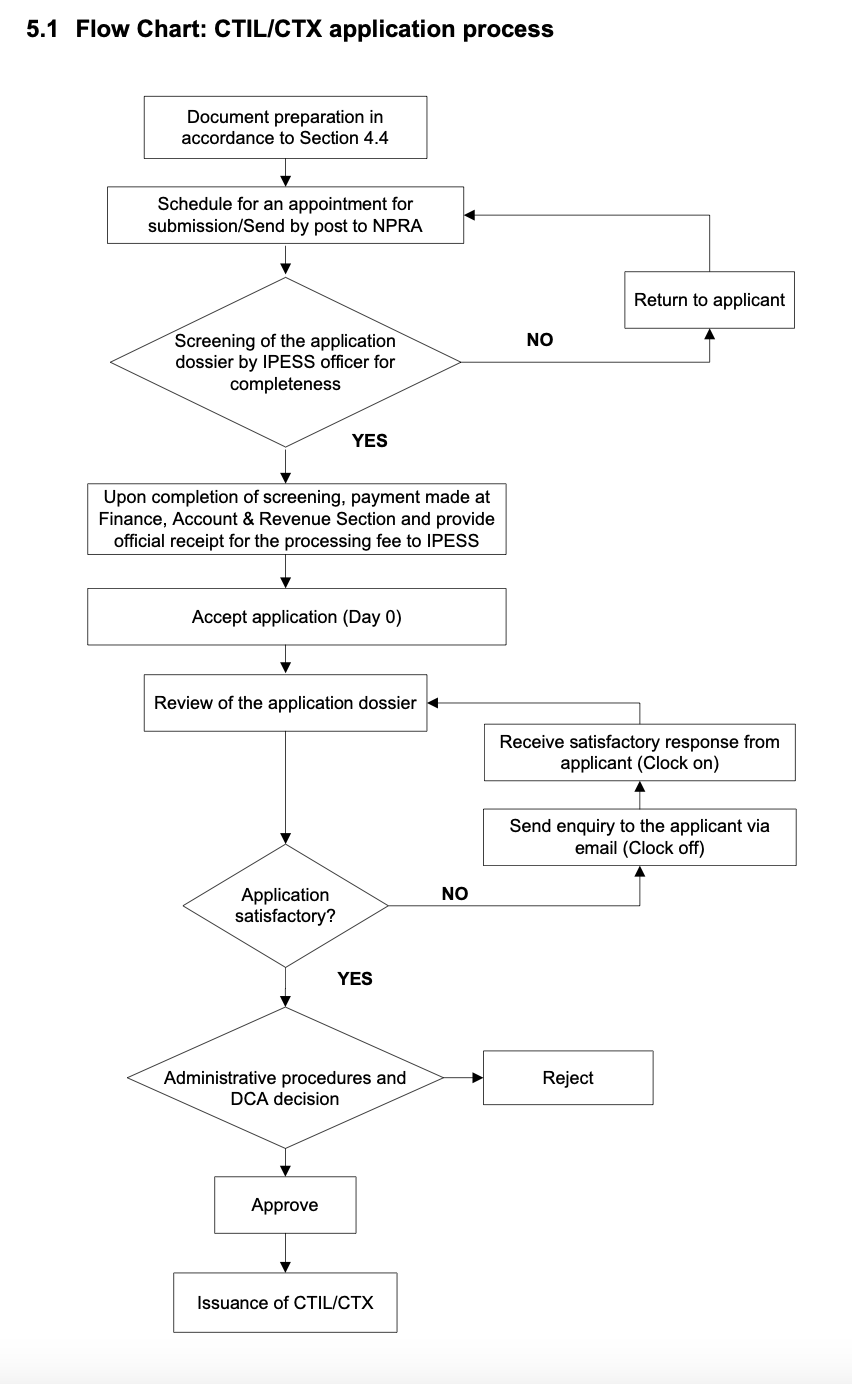
4.6 Does the RA/CA provide written acknowledgment of the submitted application? If not, describe how the application is tracked.
The applicant receives an acknowledgment of application acceptance and the clock starts.
The applicant and second contact person, if available, shall be notified via e-mail of the result of the CTIL/CTX application.
For rejected applications, a rejection letter will be issued by DCA and sent directly to the applicant via post.
4.7 What is the relevant RA/CA fee in local currency/USD? Please provide as much information as possible (e.g. if the fee is different for notification/approval, initial submission, amendment, study with IMPD and study without IMPD, etc.)
The application fee is RM 500 per product (~ $110 USD).
4.8 Does RA/CA accept checks or can payment be made electronically? Please provide details on (1) A/C number; (2) A/C Name; (3) Sort Code; (4) Swift Code; (5) Bank address, etc. Where can remittance advice notices be sent?
The processing fee shall be paid in the form of a credit card/bank draft/money order/postal order payable to 'Biro Pengawalan Farmaseutikal Kebangsaan'. Alternatively, the payment can be made using a credit card at the Finance, Account & Revenue Section.
Note: Foreign currencies are not acceptable. The processing fee is not refundable.
Application for CTX is free of charge.
4.9 Is there any guidance tool available for making electronic applications? If yes, provide the link and/or step-by-step instructions.
Yes.
4.10 Does RA/CA require any screenshots/mock screens for participant-facing materials?
Yes. Where patient-facing materials are in electronic format, screenshots should be provided.
Was this article helpful?