4. Regulatory Authority (RA)/ Competent Authority (CA)
- 7 Mins to read
- DarkLight
4. Regulatory Authority (RA)/ Competent Authority (CA)
- 7 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
4.1 Provide a list of documents needed to be submitted for RA/CA review and approval including the number of copies and/or translations as relevant.
The following electronic documents are required to be submitted via CTIS (the EU’s Clinical Trials Information System).
The EU Regulation 536/2014 specifies that the documents to be evaluated are divided into two sections:
- Part 1 (including protocol and investigator information, benefit-risk assessment; see Article 6)
- Part 2 (including requirements for consent; recruitment; suitability of the trial site as well as persons involved in the conduct; data protection; insurance; and handling of biomaterials; see Article 7)
Those are submitted via the CTIS portal (the EU’s Clinical Trials Information System):
Part I Scientific and Medicinal Product Documentation | Part II National and Patient Level Documentation |
Application Form (Italian or English) | Informed Consent form and subject information leaflet (Italian)(template available on the Ethics Committees Coordination Centre) |
Cover Letter (including sponsor’s justification for the classification as a low intervention CT, if applicable) (in English) | Recruitment arrangements and any participant-facing recruitment material (in Italian) |
Protocol and Protocol Synopsis (Italian and English) (sponsor should also consider submitting a protocol synopsis for laypersons) | Compensation arrangements (template available on the Ethics Committees Coordination Centre) |
Investigator Brochure | Suitability of investigators and facilities and Financial arrangements (Italian or English) |
Good Manufacturing Practice (GMP) documentation | Insurance/ Indemnification (in Italian) |
Investigational Medicinal Product Dossier (IMPD) / Auxiliary Medicinal Product Dossier (AMPD) | National Requirements for Data Protection |
Scientific Advice | Proof of payment |
EU Pediatric Investigation Plan (PIP) decision | Use of Biological Samples (as applicable) (template available on the Ethics Committees Coordination Centre) |
Example of IMP/AMPD labels (in Italian) |
|
4.2 Time required for RA/CA review and approval process and turnaround time if any query is raised during the review process.
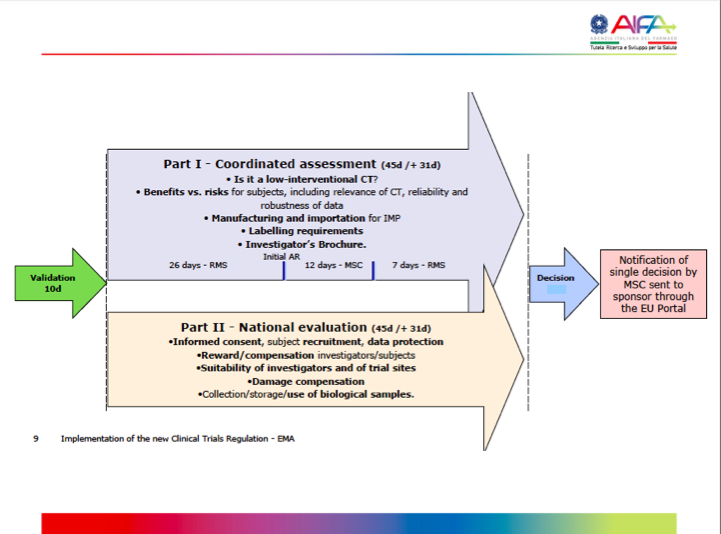
AIFA created a presentation that provides the following timelines as a guide. Further, the timelines are set out in the table thereafter, based on Art 5 (Application), Art 6 (Assessment report – aspects covered by Part I) & Art 7 (Assessment report – aspects covered by Part II), Clinical Trial Regulation (EU) 536/2014.
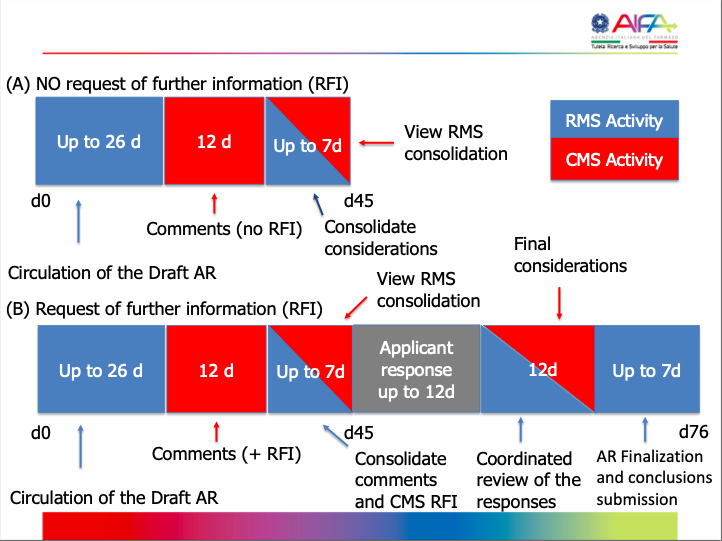
Schematic overview of timelines for an initial application:
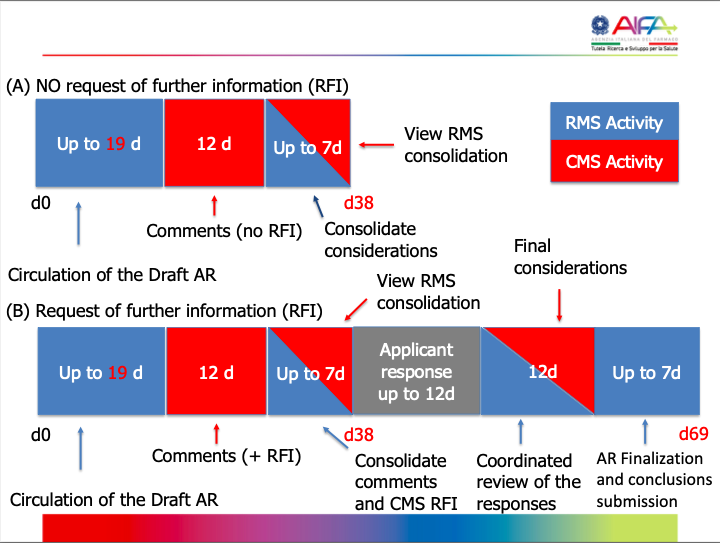
Schematic overview of timelines for a substantial modification application:
Timelines based on Art 5 (Application), Art 6 (Assessment report – aspects covered by Part I) & Art 7 (Assessment report – aspects covered by Part II), Clinical Trial Regulation (EU) 536/2014:
Validation | Part I Assessment | Part II Assessment | Decision | Total Time | Outcome |
Initial CTA | |||||
10 days (+10 / +5)
| 45 days (+12 / +19) RMS
| Same as Part I MSC | 5 days | 60 - 65 days (max. 106 days) | Authorized; Authorized subject to conditions; Rejected |
Additional MS | |||||
N/A | 10 days (+10 / +5) RMS | Same as Part I MSC | N/A | 52 days (max. 106 days) | Authorized; Authorized subject to conditions; Rejected |
Substantial Modification | |||||
6 days (+10 / +5)
| 38 days (+10 / +5) RMS
| Same as Part I MSC | 5 days (Pt I) N/A (Pt II)
| 44/49 days (Max. 90/95 days) | Authorized; Authorized subject to conditions; Rejected |
- In red: Time for sponsor to respond to questions (RFIs).
- In green: Time for RMS or MSC to assess and discuss responses.
- RMS: Reporting Member State
- MSC: Member State Concerned
If the response to the RFI is not submitted within the timeframe provided, the application will lapse.
4.3 Does the regulation support electronic submission?
Yes. All documents are to be submitted electronically through the EMA CTIS portal.
4.4 Does the regulation require the applicant to be a Principal Investigator (PI)/Chief Investigator (CI)?
No. Submission of clinical trials as per the EU CTR 536/2014 can be done by the Sponsor or authorized representative.
4.5 Please describe the process of RA/CA submission for clinical trial approval.
From 31 January 2023, all clinical trial sponsors must submit new clinical trial applications in the EU and EEA through CTIS, in terms of the EU CTR 536/2014.
A single trial application form and supporting dossier must be submitted through CTIS - a dedicated portal developed for the management of all trials in Europe (Clinical Trials Information System “CTIS”). The authorization and supervision of clinical trials will remain the responsibility of the Member States of the EU, while the EMA will manage the CTIS and the publication of its contents in the public section of the portal.





4.6 Does the RA/CA provide written acknowledgment of the submitted application? If not, describe how the application is tracked.
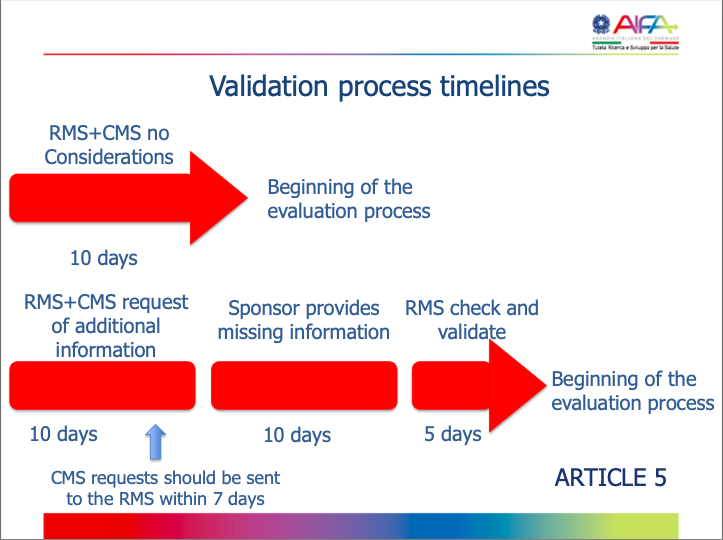
Yes, the RA/CA provides a written acknowledgment (via the CTIS portal) of the submitted application within 6 days of submission of the application.
Art 5.1, Clinical Trial Regulation (EU) 536/2014.
Tracking is via CTIS.
4.7 What is the relevant RA/CA fee in local currency/USD? Please provide as much information as possible (e.g. if the fee is different for notification/approval, initial submission, amendment, study with IMPD and study without IMPD, etc.)
There are two different fees that must be paid to AIFA for the authorization of clinical trials:
1. AIFA: With reference to the Ministerial Decree of 30 January 2023 on "Determination of the single fee for clinical trials, of the attendance fee and reimbursement of expenses for participation in meetings of the National Coordination Centre of local ethics committees for clinical trials on medicinal products for human use and medical devices, of local ethics committees and of ethics committees of national relevance"(hereinafter the "Single Fee Decree"), published on 7 February 2023 on the Official Journal, General Series no. 31, and entered into force on 22 February 2023, the following operational instructions are provided:
- As of 22 February 2023, the new fees established by the Single Fee Decree shall apply to applications for authorization of substantial amendments to clinical trials submitted through OsSC and applications for authorization of clinical trials and related substantial modifications submitted through CTIS.
- The fees identified in Annex 1 of the Single Fee Decree must be paid through AIFA's Online Payment System (POL) by means of a single payment according to the instructions contained in the POL invoice that the system will generate, with the same methods applied until now (see also communication of 13 January 2023 accessible from the "related links box").
The POL system has been updated with the specific fees indicated in the aforementioned Annex 1, which the sponsor/applicant must select based on the applicable situation. The POL system handles all fees relating to applications for authorization of clinical trials (and substantial modifications thereto) of all trial phases.
According to Annex 1 of the “Single Fee Decree”, fees include:
- Phase 1: €20.000
- Phases II and III:
- For studies with 1 to 15 sites: €17.000
- For studies with more than 15 sites: €19.000
For more on fees, please refer to Annex 1 of the Single Fee Decree.
Applicants and Sponsors are reminded of the obligation to pay stamp duty on all applications for authorization of clinical trials and related substantial amendments/modifications, submitted through the National Observatory on Clinical Trials of Medicinal Products as well as through the European CTIS Portal.
The tax is payable at a flat rate of €16.00, regardless of the size of the document transmitted.
For information on how to do this, please refer to the communications that can be reached from the "Related links" box.
The declaration of payment of stamp duty must be included in the cover letter of the application: in the case of submission of the application through the EU CTIS Portal, the relevant declaration must be included at the bottom of the indication of the Ethics Committee responsible for the evaluation.
4.8 Does RA/CA accept checks or can payment be made electronically? Please provide details on (1) A/C number; (2) A/C Name; (3) Sort Code; (4) Swift Code; (5) Bank address, etc. Where can remittance advice notices be sent?
Fees are paid electronically via the AIFA “Fee payment system (PagoPa)”. However, payment by bank transfer can be considered as an extraordinary solution only by persons unable to use the ordinary PagoPa system (subjects not resident in Italy or not holders of current accounts with banks affiliated with the PagoPA).
In such cases, bank details for electronic payments are indicated under “Systema Versamento Tariffe-DM. 6/12/2016)”.
4.9 Is there any guidance tool available for making an electronic application? If yes, provide the link and/or step-by-step instructions.
The Clinical Trial application is made by the centralized EMA CTIS portal.
Sponsors wishing to use CTIS must have an EMA account. Users who do not have an EMA account can register through the EMA Account Management facility.
Organizations may need to go through additional registration steps based on the user management approach that was used for CTIS. The organization-centered approach enables user management by an administrator at the organization level, rather than at the level of an individual trial. This is intended for organizations that run various trials through CTIS. To make use of this organization-based approach, organizations must ensure that they are registered in the EMA Organization Management System (OMS) and must register a CTIS High-Level Administrator through the EMA Account Management webpage.
Below are some guidance and training modules made available by EMA:
- Getting started with CTIS – Sponsor Quickguide
- CTIS Sponsor handbook
- Reference Materials for Clinical Trial Sponsors
4.10 Does RA/CA require any screenshots/mock screens for participant-facing materials?
Not under Part I of the application.
Participant-facing material is only submitted within Part II of the application unless participant-facing material is linked to the endpoints of the clinical trial, and those shall be provided together with the protocol in Part I of the clinical trial application. However, there is no indication as to how those documents should be provided.
Was this article helpful?