4. Regulatory Authority (RA)/ Competent Authority (CA)
- 14 Mins to read
- DarkLight
4. Regulatory Authority (RA)/ Competent Authority (CA)
- 14 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
4.1 Provide a list of documents needed to be submitted for RA/CA review and approval including the number of copies and/or translations as relevant.
Regulatory Authority Requirements
As specified in the MHRA's “Clinical trials for medicines: apply for authorization in the UK”, a clinical trial submission package to the MHRA should contain the following documents:
- Cover letter (when applicable, the subject line should state that the submission is for a Phase I trial and is eligible for a shortened assessment time, or whether it is submitted as part of the notification scheme); this letter should clearly highlight the Purchase Order (PO) number to help MHRA invoice and allocate payments promptly and efficiently.
- Clinical trial application form in PDF and XML versions
- Protocol
- Investigator’s brochure (IB)
- An investigational medical product dossier (IMPD) or a simplified IMPD
- Summary of scientific advice obtained from the MHRA or any other regulatory authority (if available).
- Manufacturer’s authorization, including the importer’s authorization and Qualified Person declaration on good manufacturing practice for each manufacturing site, if the product is manufactured outside the European Union (EU) (See MHRA's “Importing IMP products into Great Britain from approved countries” and the Manufacturing & Import section for more information).
- Copy of the United Kingdom (UK) or the European Medicines Agency’s decision on the pediatric investigation plan (PIP) and the opinion of the pediatric committee (if applicable).
- Content of the labeling of the investigational medicinal product (IMP) (or justification for its absence).
The MHRA assesses the safety and scientific value of the clinical trial, and the pharmaceutical quality of the medicinal product, ensuring that the safety monitoring, reporting, and participant follow-up measures are appropriate for the trial. The MHRA will also inspect organizations that conduct clinical trials to verify that they are conducted in line with the appropriate standards (referred to as Good Clinical Practice).
- Preparation of the Clinical Trials Application (CTA) and REC submission should not be significantly different for DCTs compared with traditional trials.
- The submission process itself will not change with DCTs, but it is important that the submission package clearly outlines how the trial will be conducted, including DCT methods such as telemedicine, wearable technologies, and eConsent.
- The sponsor should make sure that all relevant parties are involved in the submission and that all aspects of the DCT are made available to the ethics committee.
- The protocol should be transparent about what is being done remotely and who is involved in remote/decentralized activities.
- A protocol can have a statement saying: 'assessments may be performed remotely or at a location of the participants choosing, on the approval of the Investigator'.
- Any differences between an electronic document and a paper version of the same document should be explained.
- Data collection, storage, and access documentation should describe relevant safety and personal data protection safeguards, including methods for patient identification when remote data is being collected.
- The sponsor should provide a sample of the final product of approved materials once the system is ready to go live. This will include copies of the final production video for informed consent as well as a recorded demo of the data collection tools.
Tips:
- Sponsors should ensure that any scripts are adapted to UK English, for example, for emergency services in the UK, '999' is used instead of '911'.
- Trial execution and monitoring - much of the trial management, safety monitoring, and progress monitoring activities during the execution of the trial (i.e., the “clinical” phase) are similar for DCTs compared with traditional trials.
The IRAS portal includes a list of documentation to submit for a combined review of your application. The following provides some further guidance on the content of some of the specific MHRA documents:
- Cover letter: when applicable, the subject line should state that the submission is for a "Phase I healthy volunteer trial" and is eligible for a shortened assessment time, or if a risk-proportionate approach to the conduct of the study is being applied. The cover letter should clearly highlight your Purchase Order (PO) number; this will help MHRA to invoice and allocate your payments promptly and efficiently.
- Investigational medical product dossier (IMPD): note that an active substance master file (ASMF) is NOT acceptable as a substitute for an IMPD.
- Manufacturer’s authorization, including the importer’s authorization and Qualified Person (QP) declaration on good manufacturing practice for each manufacturing site, if the product is manufactured outside the EU. Please refer to further guidance covering this area.
- Content of the labeling of the investigational medicinal product (IMP): where this has not been provided, a justification for its absence will need to be provided.
Note:
- Clinical trial application form in PDF and XML versions is not required for combined ways of working (which is for Combined CTIMP or Combined CTIMP + device project on a new part of IRAS). All other types of research should be submitted as usual by the standard part of IRAS (not the new part) and it includes device-only studies, existing studies approved through standard IRAS, and Observational Studies.
- CTIMP applications via combined review should be started and submitted using the new part of the Integrated Research Application System (IRAS) and NOT in the standard part of IRAS. For Combined review applications, please refer to the Health Research Authority website.
All documents must have copy-and-paste functionality. MHRA does not currently accept password-protected documents. Other published guidance remains relevant and should be consulted for further information on the submission requirements (with consideration of the MHRA as a sovereign regulator).
If you are using an in-vitro diagnostic device in your trial, the covering letter and/or protocol should confirm that any applicable CE marking requirements have been complied with (or will be complied with prior to the study start). (Please note that Medical devices currently sold in the UK can still display the CE mark, something that has been a requirement under EU law since 1985, but medical device companies with products on the UK market now need to plan for how they will obtain a UK Conformity Assessment (UKCA mark).
4.2 Time required for RA/CA review and approval process and turnaround time if any query is raised during the review process.
Visual graphic describing the initial process and timeline:
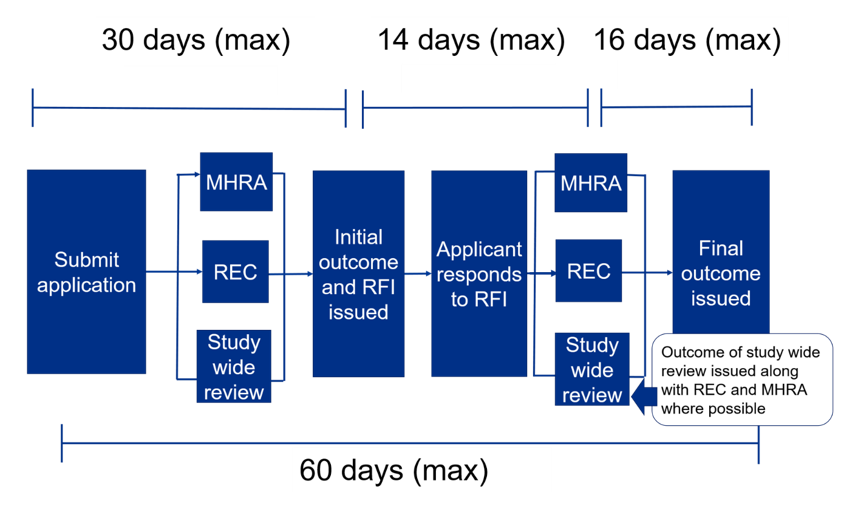
Applications for healthy volunteer trials and sponsor-determined phase I trials in non-oncology patients may qualify for a shortened assessment time and MHRA will work with the research ethics committee to endeavor to expedite these applications. Sponsors must state on the submission's cover letter if they think their trial is eligible.
Note: Trial designs that stretch to investigating the benefit of the treatment to participants may not be eligible for the expedited assessment timeframe.
MHRA will provide the outcome of the submission along with the outcome of the research ethics committee review, via the combined review process, which could be:
- Acceptance of the request for a clinical trial authorization;
- Acceptance of the request for a clinical trial authorization subject to conditions
- Grounds for non-acceptance of the request for a clinical trial authorization (GNA).
If MHRA raises grounds for non-acceptance (GNA), Sponsors will have the opportunity to respond, usually within 14 days. However, this may be extended on request.
Communication informing the applicant of the MHRA and REC decision following receipt of the responses will usually be sent within 60 days of MHRA receiving the original valid application. If an extension to the response date has been agreed on, this will impact the final decision timeline.
Notification of the decision relating to gene therapy, somatic cell therapy (including xenogenic cell therapy) products, tissue-engineered products, or products containing genetically modified organisms (Regulation 19 of SI1031) will be sent within 90 days of MHRA receiving the original application unless otherwise advised.
Timeline for a combined review assessment
The initial combined review assessment will be completed within 30 days of being submitted. In effect, it is within 30 days of being validated. If you cannot get a REC meeting date because they are all full, the 30-day clock actually does not start until 2 weeks prior to the REC meeting. IRAS is used for the initial application and supports the trial through amendments right up to the end of the trial.
- The user submits the application for coordinated review by REC, MHRA, and study-wide review and receives a response or request for further information within 30 days.
- The user responds to a request for further information within 14 days.
- REC and MHRA provide final responses within 16 days.
- The maximum time from submission to outcome from Ethics and MHRA is 60 days unless the user asks for more time. In effect, it is from validation of submission to outcome.
- Study-wide review is usually issued at the same time as REC and MHRA but may come later if there are still issues to be discussed with the user.
- If there are questions during the review, a ‘Request for Further Information’ (RFI) may be raised, and applicants will need to provide additional information or clarifications. Applicants can request an extension if more than 14 days are needed to respond to the RFI, by emailing the MHRA at clintrialhelpline@mhra.gov.uk. If more than 14 days are needed, this will affect the maximum 60-day timeline.
4.3 Does the regulation support electronic submission?
MHRA has moved from paper to automated electronic communication. To ensure that you receive all the correspondence, please ensure that you add MHRA_CT_Ecomms@mhra.gov.uk to your safe sender email list. MHRA will only send official correspondence to the named applicant's email address.
4.4 Does the regulation require the applicant to be a Principal Investigator (PI)/Chief Investigator (CI)?
In the case of commercial research, the PI/CI should be independent of the Sponsor and not directly employed by them.
The Chief Investigator (CI) is the overall lead researcher for a research project (outside the UK, the term Coordinating Investigator or Investigator may be used). In addition to their responsibilities, if they are members of a research team, chief investigators are responsible for the overall conduct of a research project. The chief investigator’s responsibilities are set out in more detail in the UK Policy Framework for Health and Social Care Research.
The Principal Investigator (PI) is an individual responsible for the conduct of the research at a research site. There should be one PI for each research site. In the case of a single-site study, the CI and PI will normally be the same person.
The UK Policy Framework does not require a UK legal representative of the lead Sponsor (or any co-sponsor) for studies other than CTIMPs. The Sponsor is the individual, organization, or partnership that takes on overall responsibility for proportionate, effective arrangements being in place to set up, run, and report a research project. All health and social care research has a Sponsor. The Sponsor is usually expected to be the employer of the chief investigator in the case of non-commercial research, or the funder in the case of commercial research. (The employer or funder is not automatically the Sponsor; they explicitly accept the responsibilities of being the Sponsor.) The Sponsor has overall responsibility for the research. Sponsors of clinical trials of investigational medicinal products have particular legal duties – see the HRA Planning and Improving Research page for details.
A Sponsor may delegate activities to a CRO, but the ultimate responsibility (e.g. for the quality and integrity of the research data) always resides with the Sponsor. This does not prevent appropriate CROs from acting as the Sponsor’s legal representative – see the HRA Planning and Improving Research page. The CRO is responsible for implementing quality assurance and quality control with respect to the activities delegated to it. Any activity that is delegated to and assumed by a CRO should be specified in writing. Any activity not specifically delegated to and assumed by a CRO is retained by the Sponsor.
4.5 Please describe the process of RA/CA submission for clinical trial approval.
Submission process:
- Full step-by-step description (also refer to Section 3.7 of this guidebook).
- Video: HRA produced a short explainer video for newcomers to combined reviews.
- IRAS Development Q&A outlines how the new UK combined review process is different.
- First, because applicants must submit to the EC and the MHRA together, not separately, the combined review may require organizations to change their operations. For example, individuals can view and revise the application information in IRAS instead of generating a PDF of an in-progress application. This new feature may affect quality control processes within the system. For example, collaborators can be granted view-only or read-write access to IRAS.
- Secondly, another change is that messages from the IRAS will only go to a specific set of contacts. If organizations have different teams working on different parts of the application, there must be effective internal communication mechanisms in place.
- IRAS messages could include Requests for Further Information (RFIs) or a final outcome issued. Finally, applicants will receive a consolidated initial outcome.
- If there is anything that needs to be addressed for the trial to be approved, applicants should respond within 14 days.
- If more time is needed to respond to an RFI, an extension can be formally requested by emailing clintrialhelpline@mhra.gov.uk and indicating how much time is needed.
- Under combined review, applicants receive a final decision on their application within 10 days of receipt of a response to an RFI.
To submit a new clinical trial application in the combined review section of IRAS, applicants must complete pre-submission procedures that include creating an account in IRAS (if a new user), creating a new project and key roles, entering and uploading project details, and routing the application to the sponsor for their review and authorization. Once the sponsor or its delegate has authorized the submission, the chief investigator (CI) or project deputy will receive a notification to complete the EC booking. When selecting an EC meeting that is not the first available meeting, the 60-day regulatory clock for both the EC and the MHRA will start on the cut-off date for the meeting that is chosen. Once booked, the EC booking page will update to show the confirmed booking details. The CI/project deputy will then be able to scroll down the page to select the option to “submit to the regulators.” See the step-by-step guide for combined review for detailed step-by-step instructions.
Other regulatory information aside from new clinical trial applications are to be submitted pursuant to the guidance to make submissions to MHRA. These submittals may include substantial amendments for existing clinical trials, end-of-trial notifications, and developmental safety update reports (DSURs).
4.6 Does the RA/CA provide written acknowledgment of the submitted application? If not, describe how the application is tracked.
The application will be validated within 3 days of being received and you will be able to see the validation status in the dashboard. Validation will check whether all of the required documents have been submitted, can be opened, and are readable. If your application is invalidated, you will need to re-submit the application.
4.7 What is the relevant RA/CA fee in local currency/USD? Please provide as much information as possible (e.g. if the fee is different for notification/approval, initial submission, amendment, study with IMPD and study without IMPD, etc.)
The sponsor or his/her designated representative is responsible for paying a fee to the Medicines and Healthcare Products Regulatory Agency (MHRA) to submit a clinical trial application for authorization. According to the MHRA Guidance to make a payment, applicants will receive an invoice to make a payment for the outstanding amount after validation of the application. Applicants must pay invoices upon receipt or they will incur penalty fees.
The MHRA updates the fees frequently; therefore, it is advised to always check on the MHRA website what are the latest fees applicable.
- £3366 British Pounds – Applications with an Investigational Medicinal Product (IMP) dossier
- £248 British Pounds – Applications without an IMP dossier
- £248 British Pounds – Clinical trial variation/amendment
- No cost – Phase IV notification
- There are no fees required for applications submitted and authorized under the Notification Scheme. However, if the MHRA raises an objection to the notification, the submission is treated as a standard request for authorization and an assessment of the submission is carried out with associated fees.
N.B.: Ethics committees (ECs) are not permitted to charge an application fee or seek any other financial contribution or donation for reviewing research proposals. Additionally, EC members receive no payment for contributing to the application review process at scheduled meetings or for attending these meetings.
4.8 Does RA/CA accept checks or can payment be made electronically? Please provide details on (1) A/C number; (2) A/C Name; (3) Sort Code; (4) Swift Code; (5) Bank address, etc. Where can remittance advice notices be sent?
According to the MHRA Guidance - Make a Payment, MHRA does not accept cheques. Payments can be made electronically by bank transfer, credit card, or debit card. Bank transfers should be sent to:
- Account Name: MHRA
- Account Number: 10004386
- Sort code: 60-70-80
- Swift code: NWBKGB2L
- IBAN: GB68NWBK60708010004386
- Bank: National Westminster Bank
- Bank address: National Westminster Bank RBS, London Corporate Service Centre, 2nd Floor, 280 Bishopsgate, London, EC2M 4RB, UK
Credit or debit card payments may be made using MHRA’s online payments service referred to as “GOV.UK Pay” in the MHRA Guidance Make a Payment.
Remittance advice notices can be sent to sales.invoices@mhra.gov.uk and should include the relevant invoice number on the remittance advice. MHRA cannot accept any documentation sent by postal mail service. Further information can be obtained by emailing sales.invoices@mhra.gov.uk. MHRA further provides that invoice disputes/queries should be emailed to Clinical Trial Applications (CTA invoices) at ctdhelpline@mhra.gov.uk and cc: sales.invoices@mhra.gov.uk.
4.9 Is there any guidance tool available for making an electronic application? If yes, provide the link and/or step-by-step instructions.
Link to full step-by-step process here.
HRA also produced a short explainer video for newcomers to combined review.
4.10 Does RA/CA require any screenshots/mock screens for participant-facing materials?
Participant-facing documents are not reviewed by the MHRA but by the REC. Please refer to Section 3.11 of this guidebook where this topic is discussed.
Was this article helpful?