3. Ethics Committee (EC)/Institutional Review Board (IRB)
- 4 Mins to read
- DarkLight
3. Ethics Committee (EC)/Institutional Review Board (IRB)
- 4 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
3.1 Provide a list of documents needed to be submitted for EC/IRB review and approval including the number of copies and/or translations as relevant.
CONEP requires applicants to submit the following documentation online:
- Cover Sheet for Research Involving Human Beings.
- Clinical research protocol (in Portuguese).
- Background, justification, and registration in the country of origin for drug and device health products.
- Description of materials, methods, rationale, expected results, and bibliography.
- Critical risk and benefit analysis.
- Duration.
- Responsibilities of researcher, institution, and sponsor.
- Criteria for project suspension or termination.
- Location of implementation of various project steps.
- Necessary infrastructure and agreement of the institution.
- Statement of Commitment from the Principal Investigator (PI).
- Informed consent form (ICF).
- Detailed research financial budget and researcher remuneration.
- Ownership of information.
- Characteristics of the participant population and justification for the use of vulnerable groups.
- Number of participants locally and globally (multicenter).
- Description of methods that affect research participants.
- Sources of material and details of the specific collection.
- Recruitment plans, inclusion, and exclusion criteria.
- PI/investigator(s) Curriculum Vitaes (CVs).
- Research project schedule.
- Foreign Research or Foreign Cooperation documentation (commitments and advantages for research participants and the country; identification of the national researcher and co-responsible institution; EC approval document in the country of origin or justification; response to the need for personnel training in Brazil; and lists of participating centers abroad and in Brazil).
- Research with new drug, vaccine, and diagnostic test document requirements (current clinical trial phase and demonstration of compliance with previous clinical trial phases; drug substance registration in the country of origin and status of research; IB; clinical information from previous trial phases; justification for using placebo or wash out period; access to the drug, if its superiority is proven; researcher's statement, justification for inclusion of healthy participants; forms of recruitment).
https://bvsms.saude.gov.br/bvs/publicacoes/manual_operacional_comites_pesquisa_4ed.pdf
http://conselho.saude.gov.br/arquivos/NO_01-12_english.pdf
3.2 Time required for EC/IRB review and approval process and turnaround time if any query is raised during the review process.
The institutional EC (CEP) is required to issue an initial report within 30 days from the date the principal investigator (PI) submits an application for review.
The CEP’s review of the protocol documentation for completeness should be accomplished within 10 days following submission. The review period must be counted from the date the project entered “Ethical Assessment” (i.e., after going through the validation of documents which takes around 10 days, and when the Certificate of Presentation for Ethical Assessment (CAAE) is issued).
If the project needs to be reviewed by CONEP, the deadline is 15 days for document validation and 45 days for ethical assessment.
3.3 Does EC/IRB have any fast-track or expedited review process?
No. There is no fast-track process.
3.4 Does EC/IRB need to be registered and/or accredited/approved by RA/CA?
Yes. The National Commission for Ethics in Research (Comissão Nacionalde Éticaem Pesquisa) (CONEP) is the central statutory body responsible for the registration, audit, and accreditation of Institutional ethics committees (ECs), known as (Committees of Ethics in Research (Comitês de Ética em Pesquisas) (CEPs) and is the advisory body for the Ministry of Health (MOH).
3.5 How frequently do EC/IRB meet?
This varies depending on the committee. Most committees meet at least monthly.
https://www.fsp.usp.br/site/pesquisa/mostra/895
3.6 Is any additional approval required apart from the EC/IRB (e.g. scientific committee, subject matter expert committee, etc.)?
For studies led by Brazilian researchers, there are no further requirements.
For trials led by foreign researchers, approval is also required from CONEP, in addition to local ethics approval.
3.7 Please describe the process of the EC/IRB submission for clinical trial approval.
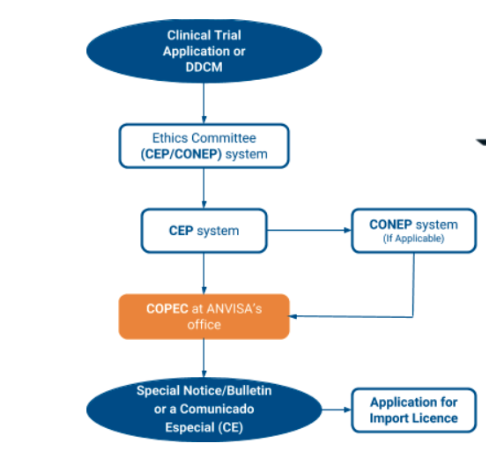
3.8 What is the relevant EC/IRB fee in local currency/USD? (e.g., is the fee different for notification/approval, initial submission, amendment, study with IMPD and study without IMPD, investigator site addition, etc.)?
CONEP does not permit ethics committees in Brazil to charge a fee to review clinical trial protocols.
3.9 Does EC/IRB accept checks or can payment be made electronically? Please provide details on (1) A/C number; (2) A/C Name; (3) Sort Code; (4) Swift Code; (5) Bank address, etc. Where can remittance advice notices be sent?
N/A.
3.10 Is there any guidance tool available for making electronic applications? If yes, provide the link and/or step-by-step instructions.
Please refer to the Brazil Platform Profile Guide. It is in Portuguese; an English version is not available.
3.11 Does EC/IRB require any mock screens/screenshots of participant-facing material on the app? If yes, do these need to be submitted in the local language?
Yes, where patient-facing materials are presented electronically, screenshots should be provided.
3.12 Does the EC/IRB have any template or specific element requirements on ICF and/or other participant-facing materials?
There is no specific template for the ICF in Brazil.
3.13 Is there any guidance tool on participant compensation including clinical study-related injury per local requirements?
The sponsor is responsible for providing compensation to research participants and/or their legal heirs in the event of trial-related injuries or death. The sponsor must also ensure that participants who suffer any trial-related injuries are provided with free medical treatment for such injuries. There is no specified amount of how much compensation should be provided.
http://conselho.saude.gov.br/resolucoes/2012/Reso466.pdf
3.14 Are there any specific local safety reporting requirements for clinical studies?
Please refer to Section 2.15 of this guidebook.
3.15 Does the EC/IRB require any periodic study reporting?
All periodic safety reporting is only required for ANVISA – the ethics committee does not require any periodic reporting.
Was this article helpful?