3. Ethics Committee (EC)/Institutional Review Board (IRB)
- 10 Mins to read
- DarkLight
3. Ethics Committee (EC)/Institutional Review Board (IRB)
- 10 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
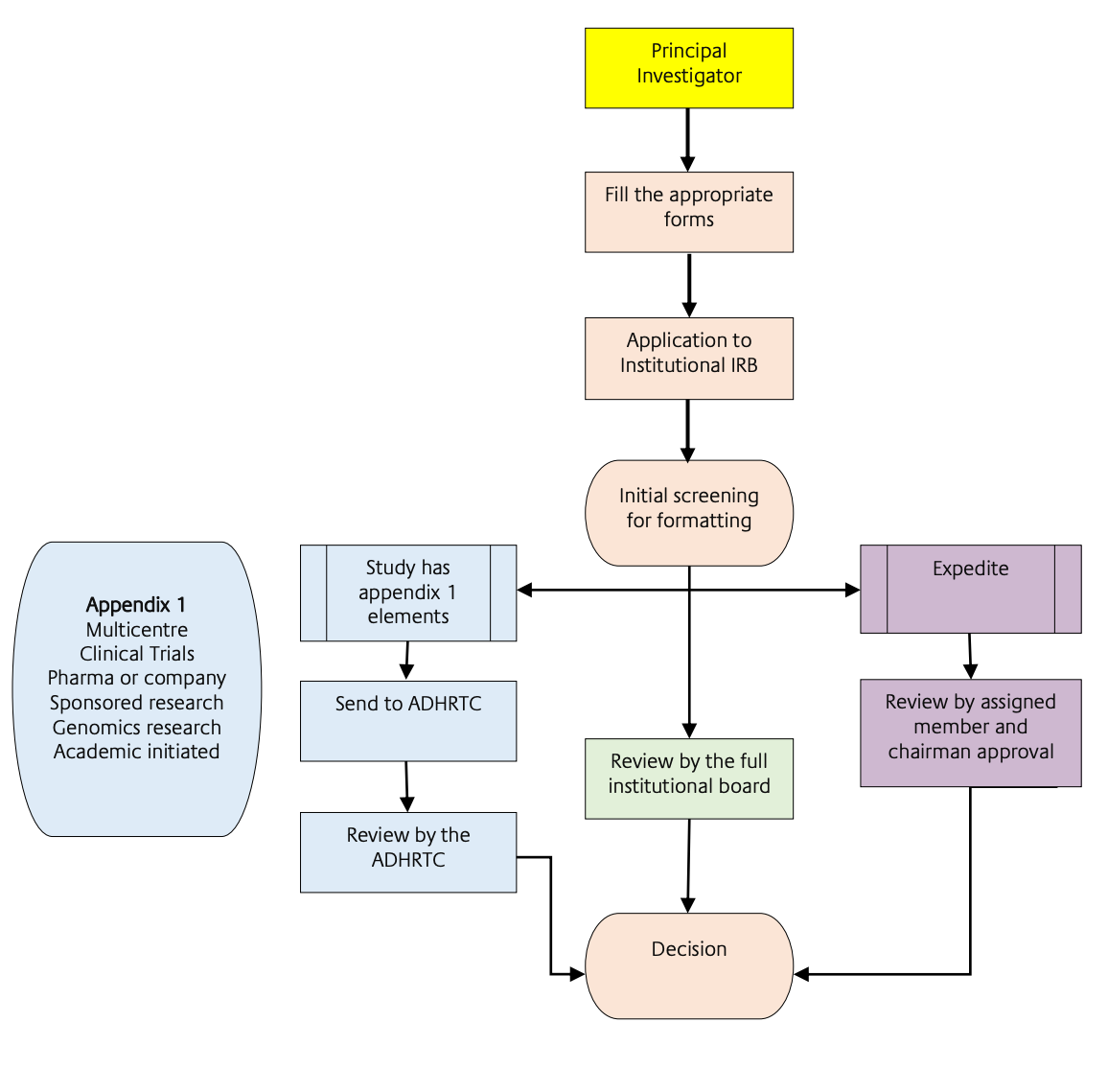
3.1 Provide a list of documents needed to be submitted for EC/IRB review and approval including the number of copies and/or translations as relevant.
The following documentation is required to be submitted to the Abu Dhabi Health Research and Technology Committee (“ADHRTC”) or REC/IRB:
- Administrative documentation
- Information about subjects
- Documentation concerning the trial protocol
- Documentation about the medicinal product tested
- Documentation about the technical requirements and the staff
- Data about funding and the administrative organization of trials
The content, format, and requirements of the documentation are set out in Appendix 2 of the DOH's “Standard on Human Subject Research”.
3.2 Time required for EC/IRB review and approval process and turnaround time if any query is raised during the review process.
The DOH’s “Guidelines for Conducting Clinical Trials with Investigational Products and Medical Devices” contain the following provisions regarding timelines for review:
“X. TIMELINES FOR REVIEW
- 32. The following timelines are as per international benchmarks:
- 31.4 Interventional Clinical Trial reviewed by ADHRTC: Within a period of 90 calendar days of filing an application, the REC concerned should rule, issuing an opinion, which should be sent to the applicant.
- 31.5 Non-interventional Clinical Trial reviewed by ADHRTC: Within a period of 45 calendar days of filing an application, the REC concerned should rule, issuing an opinion, which should be sent to the applicant.
- 31.6 Interventional Clinical Trial reviewed by REC/IRB: Within a period of 60 calendar days of filing an application, the REC/IRB concerned should rule, issuing an opinion, which should be sent to the applicant and ADHRTC.
- 31.7 Non-interventional Clinical Trial reviewed by REC/IRB: Within a period of 30 calendar days of filing an application, the REC/IRB concerned should rule, issuing an opinion, which should be sent to the applicant and ADHRTC.”
3.3 Does EC/IRB have any fast-track or expedited review process?
Yes. The DOH’s “Standard on Human Subject Research” specified the expedited REC/IRB review process in Appendix 2:
“b. Expedited
Research that poses only minimal risk to participants can be handled as Expedited. “Minimal risk” means that the probability and magnitude of harm or discomfort anticipated in the proposed research are not greater than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.
- In the case of Expedited research, the REC Chair and one other member of the REC should review the proposal and advise the principal investigator of the outcome.
- Activities approved for Expedited review include:
- Collection of biological specimens through non-invasive means; for example, electrocardiography, electroencephalography, thermography, Doppler blood flow, echocardiography, functional magnetic resonance imaging;
- Clinically routine non-invasive procedures such as muscular strength testing, moderate exercise, body composition assessment, and flexibility testing involving health subjects;
- Research on individual or group characteristics or behaviour (including but not limited to research involving perception, cognition, surveys, interviews, and focus groups) as follows:
- involving adults, where (i) the research does not involve stress to subjects; and (ii) identification of the subjects and/or their responses would not reasonably place them at risk of criminal or civil liability or be damaging to the subjects' financial standing, employability, or reputation;
- involving children, where (i) the research involves neither stress to subjects nor obtaining of sensitive information about themselves, or their family; (ii) parents/guardians will complete the usual consent form (i.e. there is no request for a waiver); (iii) identification of the subjects and/or their responses would not reasonably place them or their family members at risk of criminal or civil liability or be damaging to the financial standing, employability, or reputation of themselves or their family members;
- Collection of data from voice, video, digital, or image recordings, as long as identification of the subjects and/or their responses does not place them at risk for criminal or civil liability, or damage their financial standing, employability, or reputation;
- Research involving existing identifiable data, documents, records, or biological specimens (including pathological or diagnostic specimens), where these materials have been collected prior to the research for a purpose other than the proposed research, confidentiality should be strictly maintained, and information should not be recorded anonymously (e.g., use will be made of audio or-videotapes, names will be recorded, even if they are not directly associated with the data); (in case where, data or human subject’s biospecimens, whether collected prospectively or retrospective, banked, are shared outside the supervision of the REC, a data and material sharing agreement should be required between the parties involved with sufficient information with confidential material transfer agreement, both in Arabic and English languages). Furthermore, a Certificate of Confidentiality should be signed.
- Continuing review of non-exempt research previously approved by the REC, where no new subjects will be enrolled or where the research involves no greater than minimal risk."
3.4 Does EC/IRB need to be registered and/or accredited/approved by RA/CA?
Yes. The DOH’s “Standard on Human Subject Research” defines an REC/IRB as follows:
“3.21 Institutional Review Board (IRB): Is equivalent to the Research Ethics Committee (REC). It is an administrative body established within the facility that is intending to conduct clinical research, to protect the rights and welfare of human research subjects recruited to participate in research activities conducted under their facility, in addition to the legal protection for the investigators and the facility itself.”
“4.2 Establishing and Operating a Facility Research Ethics Committees (Facility REC) - Research Ethics Committees are authorized by DOH to ensure the protection of human subjects in research projects conducted or sponsored (see Appendix 1 on DOH Guidelines for the Structure and Function of REC) by the facility.
- 4.2.1 A healthcare provider/facility seeking DOH authorization to Conduct Human Subject Research must ensure that a Research Ethics Committee is established and maintained for each facility that holds a Facility Research Authorization.
- 4.2.2 The Research Ethics Committee must, at a minimum:
- 4.2.2.1. Include members from departments that are more active in clinical research;
- 4.2.2.2. Be multi-representative and multi-disciplinary;
- 4.2.2.3. Have clear SOPs and protocols;
- 4.2.2.4. Have protocols for different levels of research approvals;
- 4.2.3 When establishing Research Ethics Committees, facilities and investigators must comply with The Integrated Addendum to the International Conference on Harmonization Good Clinical Practice (E6) (R2), the International Standard for Ethical Research Conduct, Chapter VII of the Health Regulator Manual, and all other applicable Laws and Regulations.
- 4.2.4 The facility must ensure that the REC is constituted and operates in accordance with DOH published rules and regulation.
- 4.2.5 Before confirming a favorable opinion on any research (including both CTIMPs and non-CTIMPs), the REC shall assure itself that the sponsor and investigators have appropriate insurance or indemnity cover for the potential legal liability arising from the research due to either injury or death attributable to the research especially in the case of CTIMP.
- 4.2.6 As authorized by DOH, the Research Ethics Committee should:
- 4.2.6.1 Review the research application and ensure its compliance with national laws and standards, international research standards, and DOH regulations;
- 4.2.6.2 Give an ethical opinion on proposed research projects;
- 4.2.6.3 Refer to DOH for its approval of all proposals for proposed research in critical areas;
- 4.2.6.4 Monitor the research process and review regular reports from the PI;
- 4.2.6.5 Be notified of any serious breach of Good Clinical Practice or the research protocol;
- 4.2.6.6 Review protocol deviations for approval/rejection;
- 4.2.6.7 Communicate any change in its ethical position to DOH;
- 4.2.6.8 Notify DOH where one of the following is suspected:
- 4.2.6.8.1. Conduct of a trial without regulatory authorization or favorable REC opinion.
- 4.2.6.8.2. Provision of false or misleading information to the REC in relation to an application for ethical opinion or notification of substantial amendment.
- 4.2.6.8.3. Implementation of a substantial amendment without authorization and/or a favorable opinion as appropriate.
- 4.2.6.8.4. Failure to notify SUSARs occurring in a trial in an expedited manner or to provide an Annual Safety Report.
- 4.2.6.8.5. Failure to notify urgent safety measures.
- 4.2.6.8.6. Failure to notify the early termination or conclusion of the trial.
- 4.2.6.8.7. A serious breach of ICH GCP or the research protocol. A breach of the conditions and principles of ICH GCP or the research protocol should be regarded as “serious” if it is likely to affect to a significant degree the safety or physical or mental integrity of the participants or the scientific value of the trial.
- 4.2.6.8.8. Any other fraud or serious misconduct.
- 4.2.6.8.9. Consideration should also be given to notifying the authorities where a pattern emerges of repeated minor breaches of ICH GCP or the research protocol.”
3.5 How frequently do the EC/IRB meet?
From the research conducted, the frequency of REC/IRB meetings is not specified. However, The DOH’s “Guidelines for Conducting Clinical Trials with Investigational Products and Medical Devices” contain the following provisions:
“VI. ETHICS AND RESEARCH COMMITTEES
24. Clinical trial scope, purpose, type and design determine the appropriate body or bodies, which should be involved in the assessment of the application.
- 24.1 This includes the involvement of Ethics and Regulatory Committees within the timelines for the authorization of that clinical trial as set out in this Guideline.”
3.6 Is any additional approval required apart from the EC/IRB (e.g. scientific committee, subject matter expert committee, etc.)?
No. No additional approval is specified besides that of ADHRTC.
3.7 Please describe the process of the EC/IRB submission for clinical trial approval.
Please refer to Sections 3.1 and 3.4 above.
3.8 What is the relevant EC/IRB fee in local currency/USD? (e.g., is the fee different for notification/approval, initial submission, amendment, study with IMPD and study without IMPD, investigator site addition, etc.)?
From the research conducted, the exact REC/IRB fee could not be established.
The DOH’s “Guidelines for Conducting Clinical Trials with Investigational Products and Medical Devices” contain the following provisions regarding payment of fees:
“28.4 The IRB/REC may collect a fee for the submission of applications requesting an opinion. The fee should be in the amount determined in the tariff.”
3.9 Does EC/IRB accept checks or can payment be made electronically? Please provide details on (1) A/C number; (2) A/C Name; (3) Sort Code; (4) Swift Code; (5) Bank address, etc. Where can remittance advice notices be sent?
From the research conducted, these details could not be determined.
3.10 Is there any guidance tool available for making electronic applications? If yes, provide the link and/or step-by-step instructions.
The DOH’s “Standard on Human Subject Research” contains detailed guidance on the procedure and required documentation for submission to the ADHRTC (Appendix 1 and 2) and the REC/IRB (Appendix 3).
APPENDIX 1: Guideline on SOPs for Conducting Clinical Trials for Investigational Medicinal Products (CTIMPs).
APPENDIX 2: The following points are in specific reference to “Guidelines for Conducting Clinical Trials with Investigational Products and Medical Devices”.
APPENDIX 3: Guideline on REC/IRB Functions and Structure (illustrated by organogram).
3.11 Does EC/IRB require any mock screens/screenshots of participant-facing material on the app? If yes, do these need to be submitted in the local language?
It is advisable that the Sponsor planning to use eConsent in Dubai discuss it with the local EC/s and the DOH prior to submission. The DOH will then advise if additional documentation (i.e. screenshots) is required or not.
3.12 Does the EC/IRB have any template or specific element requirements on ICF and/or other participant-facing materials?
The DOH’s “Guidelines for Conducting Clinical Trials with Investigational Products and Medical Devices” address ICF (clauses 16- 18).
The Health Authority Abu Dhabi issued, in January 2016, “Guidelines for Patient Consent”. The purpose of this guideline is to set out principles, to advise on best practices, to provide a framework for good practice, to identify practices that support patient rights to participate in informed decision-making, to recommend measures that Health Care Facilities and to respect local custom in regards to patient consent.
The Department of Health does not currently have any ICF templates; however, at a site level, the Local EC/ IRBs may have specific ICF templates. However, Appendix 6 of the DOH “Standard on Human Subject Research” provides detailed information on the minimum requirements.
3.13 Is there any guidance tool on participant compensation including clinical study-related injury per local requirements?
The DOH “Standard on Human Subject Research” provides some guidance on compensation. This can be found under Appendix 11: Guideline on Risk and Benefit:
“Compensation is NOT a benefit:
i. Investigators may pay research subjects for their participation or offer gift certificates or vouchers.
ii. However, it is important that payments or gifts not be so ample as to coerce participation from those who might otherwise decline to be a part of the study. Payment should not encourage subjects to participate or continue to participate against their better judgment.
iii. Subjects should receive partial payment if they withdraw from a study.
iv. Withholding all payment until participation is complete is coercive. A modest lump sum can be paid after a subject’s ‟ participation is complete if the arrangement is thoroughly documented in the consent form."
Chapter 2 of Federal Law No. 8 of 2019 on Medical Products, Profession of Pharmacy and Pharmaceutical Institutions sets out provisions in relation to Clinical and Non-clinical Studies in the UAE. Article 16 requires “the entity for whose interest” a clinical study is conducted to commit to concluding “an insurance contract with any insurance company operating in the State, to cover the damage that may arise from the study.”
3.14 Are there any specific local safety reporting requirements for clinical studies?
Please refer to Section 2.15 of this guidebook.
3.15 Does the EC/IRB require any periodic study reporting?
Please refer to Section 2.16 of this guidebook.
Was this article helpful?