3. Ethics Committee (EC)/ Institutional Review Board (IRB)
- 23 Mins to read
- DarkLight
3. Ethics Committee (EC)/ Institutional Review Board (IRB)
- 23 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
3.1 Provide a list of documents needed to be submitted for EC/IRB review and approval including the number of copies and/or translations as relevant.
Submissions of clinical trial applications (CTAs) in the UK are made to the Integrated Research Application System (IRAS). The portal includes a list of documentation to submit for the combined review of the EC application.
Submission documents are uploaded to the IRAS portal. The UK has a combined review process in which submission to the MHRA and EC takes place at the same time via the IRAS portal.
The full detailed list of documents can be found on the HRA website under “Document management for combined review applications”. Below is a listing of “mandatory” documents which are just applicable to REC review.
Document Type | Mandatory or required | Review body it’s sent to |
Cover letter | Mandatory | REC |
Protocol | Mandatory | REC |
Investigator Brochure/ SmPC | Mandatory | REC |
Proof of Insurance | Mandatory | REC |
Financial arrangements | Not mandatory | REC |
Suitability of clinical trial site facilities | Not mandatory | REC |
Recruitment arrangements | When applicable | REC |
Participant Information and Informed Consent Form | Mandatory | REC |
Suitability of the Investigator/Investigator CV | Mandatory | REC |
3.2 Time required for EC/IRB review and approval process and turnaround time if any query is raised during the review process.
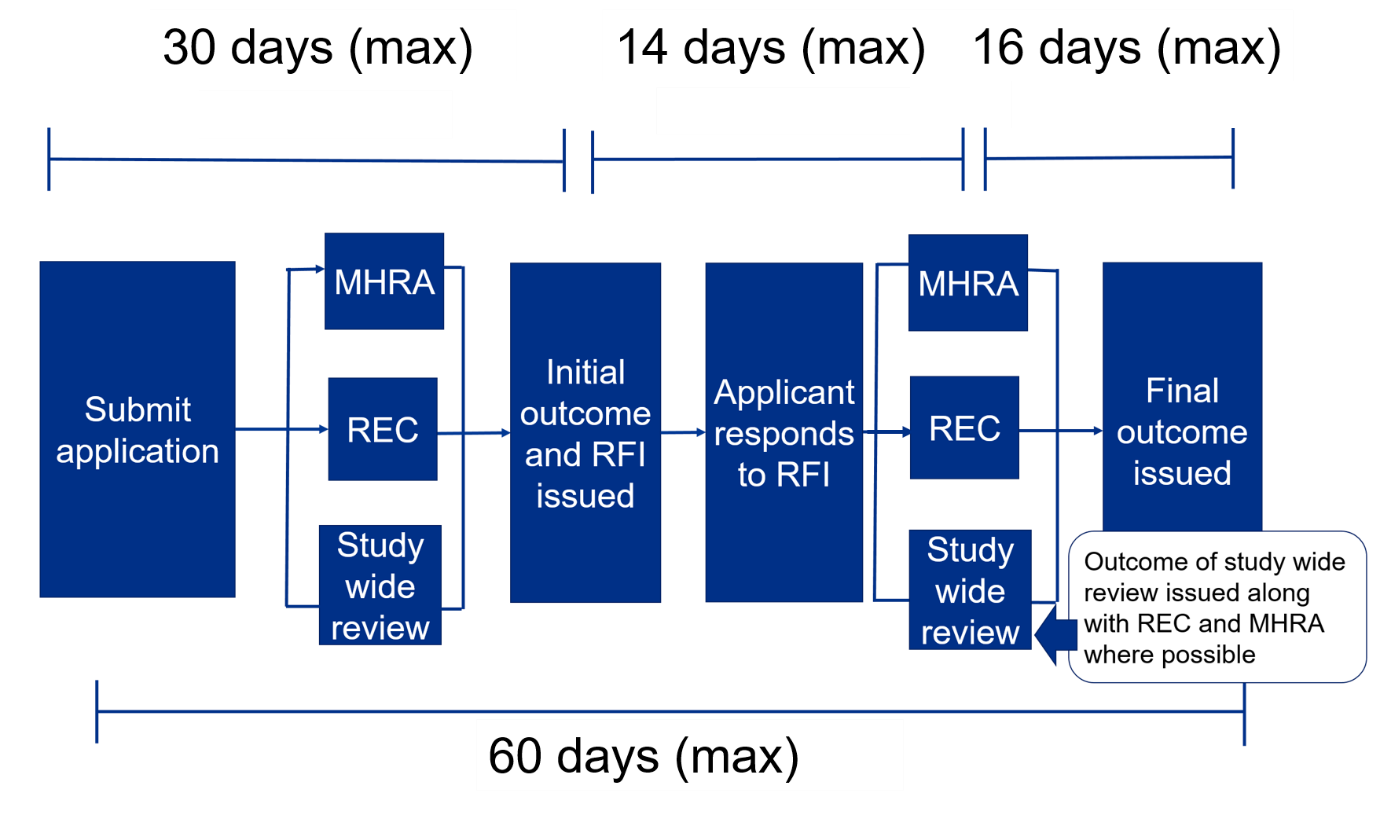
The initial combined review assessment will be completed within 30 days of being submitted. In effect, it is within 30 days of being validated. If you cannot get a REC meeting date because they are all full, the 30-day clock actually does not start until 2 weeks prior to the REC meeting. IRAS is used for the initial application and supports the trial through amendments right up to the end of the trial.
- The user submits the application for coordinated review by REC, MHRA, and study-wide review and receives a response or request for further information within 30 days.
- The user responds to request for further information within 14 days.
- REC and MHRA provide final responses within 16 days.
- The maximum time from submission to outcome from Ethics and MHRA is 60 days unless the user asks for more time. In effect, it is from validation of submission to outcome.
- Study-wide review is usually issued at the same time as REC and MHRA but may come later if there are still issues to be discussed with the user.
If there are questions during the review, a ‘Request for Further Information’ (RFI) may be raised, and applicants will need to provide additional information or clarifications. Applicants can request an extension if more than 14 days are needed to respond to the RFI, by emailing the MHRA at clintrialhelpline@mhra.gov.uk. If more than 14 days are needed, this will affect the maximum 60-day timeline.
Initial Review Process and Timelines - Health Research Authority (hra.nhs.uk)
3.3 Does EC/IRB have any fast-track or expedited review process?
Yes, the REC offers a fast-track ethics review service for research applications that need a rapid research ethics review with one of their existing RECs for global clinical and phase I trials for any disease area which will be quicker than their usual review time.
- This link leads to the page which has all the information you need about how to apply for fast-track review.
- Sponsors will need to contact one of the RECs participating in the fast-track service directly to arrange this. HRA will list these RECs on this web-page along with instructions.
- Applicant submitting via the combined review process should complete in the new part of IRAS.
- For specific queries about combined review, please visit the combined review guidance page.
3.4 Does EC/IRB need to be registered and/or accredited/approved by RA/CA?
No, in accordance with provisions of the Care Act 2014, the UK Health Research Authority (HRA) was established as an executive non-departmental public body (NDPB) sponsored by the UK Department of Health on January 1st, 2015. HRA and the Devolved Administrations provide a Research Ethics Service so that research proposals relating to their areas of responsibility can be reviewed by a Research Ethics Committee (REC).
- Managing the Research Ethics Committees in England is one of the Health Research Authority’s core functions.
- HRA has a duty to provide an efficient and robust ethics review service that maximizes UK competitiveness for health research and maximizes the return from investment in the UK while protecting participants and researchers.
- HRA has a dual mission to protect the rights, safety, dignity, and well-being of research participants and to facilitate and promote ethical research that is of potential benefit to participants, science, and society.
- Under the UK Health Department's Governance Arrangements for Research Ethics Committees (GAfREC), each Research Ethics Committee (REC) within the Research Ethics Service, is required to adopt Standard Operating Procedures (SOPs) approved by or on behalf of its appointing authority.
- The REC is required to act in accordance with its SOPs and is ultimately accountable to its appointing authority for its governance in this respect.
- 7.5.1 of the Standard Operating Procedures for Research Ethics Committees came into effect on August 2nd, 2021. The Governance Arrangements for Research Ethics Committees (GAfREC) describe what is expected from RECs when reviewing research proposals.
3.5 How frequently do EC/IRB meet?
This REC Directory page provides a list of meeting dates for Research Ethics Committees (RECs) within the UK Health Departments’ Research Ethics Service.
Please ensure you are aware of the latest guidance about applying for approvals before you plan and book for your REC review.
After preparing your application, please use the Online Booking Service to book a REC meeting.
3.6 Is any additional approval required apart from the EC/IRB (e.g. scientific committee, subject matter expert committee, etc.)?
Yes, depending on the type of studies, it may be required to involve additional committees, all of them can be done via the IRAS portal.
IRAS captures the information needed for the relevant approvals from the following review bodies:
- Administration of Radioactive Substances Advisory Committee (ARSAC)
- Confidentiality Advisory Group (CAG)
- Gene Therapy Advisory Committee (GTAC)
- Health Research Authority (HRA) and Health and Care Research Wales (HCRW) for projects seeking HRA & HCRW Approval
- Medicines and Healthcare products Regulatory Agency (MHRA)
- NHS / HSC R&D offices
- NHS / HSC Research Ethics Committees
- HM Prison and Probation Service (HMPPS)
- Social Care Research Ethics Committee
3.7 Please describe the process of the EC/IRB submission for clinical trial approval.
This step by step guide to using IRAS for combined review provides very detailed information on the steps required.
The complete application process of EC/IRB submission of CTA is described below:
A) Pre-submission
- Finalize the protocol and supporting documents.
- New users create IRAS accounts. Create a new project and allocate roles as required.
- Complete project details, study information, and clinical trial dataset in IRAS and upload supporting documentation.
- Application is sent to the sponsor assigned in step 2 to review and authorize. It also has to be signed off by the Chief Investigator within IRAS.
- Book a REC online and submit the application - note that except for Phase 1 healthy volunteers, you can no longer get a REC meeting date ahead of submission.
B) Post-submission
Application submitted for combined review by REC and MHRA:
Studies taking place in the NHS/HSC are also reviewed against NHS standards. REC review and MHRA review then take place at the same time. Either review can lead to a request for further information (RFI) (if required). The applicant submits responses to RFIs in IRAS which then feedback into each review in a circular process, or alternatively:
- REC review can lead to REC approval issued
- HRA approval will apply to NHS sites
- MHRA review can lead to MHRA approval
All of the above then lead to 'For studies taking place in the NHS/HSC study-wide review completed’.
C) Making an initial submission
Preparing your application:
As mentioned above, the following steps should be completed before you go on to create your project in IRAS for combined review:
- Make sure all of the supporting documentation is ready and files are clearly named with the date and version number.
- Read the general guidance on the combined review process found on the HRA website.
When you have completed the above steps, you can then proceed to IRAS for combined review and preparing your application.
- Go to the New Project tab and follow the options to create a new project. You will need to specify if you are the Chief Investigator, Project Deputy, or Collaborator. Once you have selected your role, you will be taken to the Project details page. This is where you start to build your project.
- Answer the questions on the Project Details page to reflect the details of your project. You need to complete all the fields on the Project Details page marked as mandatory before you can proceed. Please be aware that you no longer need to provide an EudraCT number, although if you have one you can still enter it here. If you are not providing an EudraCT number, it is important to leave this field completely blank (entering an answer such as ‘n/a’ will cause an error at the verification stage).
- To add the Sponsor or Sponsor Delegate Organization details, start typing the name of the required sponsor group organization. A blue triangle in the corner of a text box indicates that you need to select from one of the available pre-existing options. Start typing the information you need to enter and select the correct option when it appears (if you are a CRO and will be responsible for submitting the application, please ensure the CRO is set as the Sponsor or Sponsor Delegate Organization role). If you are not sure who to enter at this stage you can leave it blank – you will be able to add this information later. For your CI or Project Deputy role, do the same.
- You will need to enter a study end date – it does not matter at this stage if you do not know the exact date, please just enter a best estimate. It can be amended later if required.
- Below these questions you will find a set of project filter questions – please answer these, in line with the study scope. At the bottom of the page, click ‘Save and Continue’ to proceed. Your answers will be saved if you wish to return to your setup another time.
- Please note: once you click ‘Save and Continue’ your invitations to the CI / Project Deputy / Sponsor or Sponsor Delegate Organization are sent. Once they have been accepted, they will also have oversight of the project via their lists/tasks.
- The Sponsor or Sponsor Delegate Organization and CI will need to accept the project before you can proceed to submission. They will not be able to accept the project whilst it is open in another account, so please make sure you are not working on the project while they are accepting it.
- On the next page - Project dashboard - you will see headers for each of the sections that you now need to complete including:
- project contents
- study information
- medicines information (previously known as 'CWOW Clinical Dataset') - If you are returning to work on your project after logging out, you can access the project dashboard by selecting ‘My tasks’ – ‘My personal tasks’ – project.
- Under ‘Update project details’, you can make changes to the information you entered here earlier if required. You will not be able to change the Sponsor or Sponsor Delegate Organization if they have already accepted the project – after this, any sponsor group changes can only be made by the Sponsor or Sponsor Delegate.
- Click ‘Study information’. This will take you to a page where you provide more information about the study. Complete all questions as fully as possible. If you require further information on any of the questions, you can select the ‘contextual help with questions’ button at the top of the page, which will display guidance where available.
- When finished, at the bottom of the page click ‘Save and verify answers’. This will flag up any outstanding questions that still need completing. Once fully completed and no questions are being flagged, click ‘Update question set’ this will then show a ‘Content verified’ status.
- Click ‘Medicines Information (Previously known as CWOW Clinical Dataset’). You must complete this dataset in full in order for the application to be submitted.
- You can complete the question set in any order you prefer. When working on a section, click ‘Save and verify answers’ before navigating away from the page or logging out. This will save your responses and will also flag in red any incomplete or incompatible responses. When the page is complete and all fields under that section are verified, the section will be marked as ‘content verified’ so that you can see at a glance it is complete. You will still be able to make changes prior to submission if required.
- If you have used the EudraCT site to prepare an application (e.g. in the case of projects also taking place in EU countries where the same information will apply to the UK application), you will still need to provide Medicines Information Dataset in IRAS. To do this, click the ‘Import EudraCT XML’ button on the Project Dashboard. You can then upload an xml file of the application which will populate all data fields in the Medicines Information Dataset. You should then check through the dataset to ensure that all fields are appearing as expected and make any corrections as required, and answer any additional questions present in the Medicines Information dataset but not on the EudraCT form. Note: the short title on your IRAS project must exactly match the short/abbreviated title on the EudraCT form you are importing, or this could cause the import to fail. So if you are receiving an error message, please check the short title.
- Starting 31 January 2022, for applications using CTIS (Clinical Trials Information System) for projects also taking place in EU countries, you may record a European Union Clinical Trial number (EU CT number) under ‘Other Identifiers’ in the ‘Trial identification’ section of your IRAS application.
- ‘Project documents’ is where you will upload supporting documents. You can either drag and drop up to 15 documents at a time or browse if they are in different folders on your computer. You can add documents at any point whilst preparing your project, up until the point the application is sent to the Sponsor or Sponsor Delegate for review and authorization at which point the ability to edit the project will be locked.
- For each document you upload, you will need to assign a document type (please ensure you select the correct document type to ensure the correct documents go to the correct review body), and enter the document title, sponsor version number, and date. For this reason, it is strongly recommended that you clearly name each file with this information to make it easier for you to enter it during the upload process.
- The version number/date you enter must match the version number/date on the document. This will be part of the validation once submitted and will appear on the REC's final opinion letter. If the information does not match, your application may be rejected.
- Certain document types are mandatory, which means you will not be able to proceed to submission without uploading them.
- In the unlikely event that your document type does not match any of the drop-down options, please select ‘miscellaneous’ and select which review body the document is relevant to.
- It is not possible to go back and edit information once entered. If you make a mistake, please delete the document by clicking the dustbin icon, and re-upload.
- On the Project Dashboard, under ‘Key associates’, you will see details of the CI, Project Deputy, or any Collaborators with yourself named as your selected role, and the Sponsor or Sponsor Delegate Organizations. Each will have a requested status or a confirmed status. If the request from the CI or Project Deputy to a sponsor organization has not yet been confirmed, you have the option to withdraw the request by clicking ‘update key associates’ and selecting the appropriate option. If the role has been confirmed, this option will no longer appear. Further information on adding a Sponsor or Sponsor Delegate Organization and collaborators is found in the ‘User Roles and Tasks’ section further up this page.
- NOTE: You can download a draft application at any time before it has been submitted. To do this, from the main project dashboard select the 'actions' menu in the top right-hand corner of the screen and choose 'download draft application'. This will generate a PDF containing the study information and medicines information question sets.
- When you think you are ready to send the application to the Sponsor or Sponsor Delegate for review, click ‘Check your answers’ from the project dashboard which will take you to a ‘Check your answers’ page. Scroll down to the bottom of the page and click ‘Identify missing information’. This will show anything outstanding via a pink banner at the top of the page – please correct flagged items as instructed. Once complete, please click ‘Identify missing information’ again and repeat until no pink banner is shown.
- When all of the above steps are complete, and you have your ‘Key Associates’ in place, ‘Project documents’ uploaded, and a ‘Content verified’ status in place next to any questions set, you can then send your application to the Sponsor or Sponsor Delegate for review and authorization, by clicking the 'Request Review' button.
- Please note: now that the project has been sent for review, you can no longer access the project via 'My tasks' - 'My personal tasks' as this dashboard only appears when you are creating or amending project information or supporting documents. You can now access the project by clicking 'My Projects' - and the project. This is a read-only view. If you wish to see the contents of the initial submission, scroll down to 'Project history' and click on 'Initial submission'. As the relevant authorizations are completed, this is where your approvals and time stamps will be shown.
- Sponsor or Sponsor Delegate Organization review and authorization. When the application has been submitted to the Sponsor or Sponsor Delegate Organization responsible for managing the submission’s account, they will find the project under 'My Tasks' – 'My Organizational Tasks', with an outstanding task to confirm submission. They can then review the completed dataset and if happy with it, check the radio button labeled ‘I confirm this submission’, followed by the ‘submit’ button. This will then generate a task in the 'My outstanding tasks' list for the user who sent the request to 'Complete the REC Booking'. The Sponsor or Sponsor Delegate providing this confirmation is equivalent to agreeing to the following:
- 'I hereby confirm that/confirm on behalf of the sponsor (delete which is not applicable) that:
- the information provided is complete;
- the attached documents contain an accurate account of the information available;
- the clinical trial will be conducted in accordance with the protocol; and
- the clinical trial will be conducted, and SUSARs and result-related information will be reported, in accordance with the applicable legislation.'
- If the Sponsor or Sponsor Delegate Organization reviews the application and wishes to request updates to the dataset before it can be submitted, they can select ‘I do not confirm this submission’ and then provide details of the required updates in the ‘Reason for not confirming’ box. Clicking the Submit button will transfer the application back so that it can be edited as necessary.
- 'I hereby confirm that/confirm on behalf of the sponsor (delete which is not applicable) that:
- Completing the REC booking. Once the Sponsor or Sponsor Delegate has confirmed the submission, the user who sent the request will receive a task in their 'My outstanding tasks' list to 'complete the REC booking'. Selecting this task will take you to the REC booking page. This page provides a list of previous bookings and instructions for booking your REC. To book using the online booking service, applicants should select 'create booking'. The REC booking page also provides instructions on what to do if you have booked your REC manually via telephone (or email). For manual bookings, it is important to remember to import the REC booking details into your project manually to link the booking with the application.
- Please note that when selecting a REC meeting that is not the first available meeting, the 60-day regulatory clock for both the REC and the MHRA will start on the cut-off date for the meeting that is chosen. The cut-off date is 14 days before the meeting date. Once booked, the REC booking page will update to show the confirmed booking details. The applicant will then be able to scroll down the page to select the option to 'submit to the regulators'. As mentioned above, you can access the read-only view of the project by clicking 'My Projects' - and the project. Scroll down to 'Project history' and click on 'Initial submission' if you wish to check the REC booking details.
3.8 What is the relevant EC/IRB fee in local currency/USD? (e.g., is the fee different for notification/approval, initial submission, amendment, study with IMPD and study without IMPD, investigator site addition, etc.)?
Ethics committees (ECs) are NOT permitted to charge an application fee or seek any other financial contribution or donation for reviewing research proposals. Additionally, EC members receive no payment for contributing to the application review process at scheduled meetings or for attending these meetings.
3.9 Does EC/IRB accept checks or can payment be made electronically? Please provide details on (1) A/C number; (2) A/C Name; (3) Sort Code; (4) Swift Code; (5) Bank address, etc. Where can remittance advice notices be sent?
Ethics committees (ECs) are NOT permitted to charge an application fee or seek any other financial contribution or donation for reviewing research proposals. Additionally, EC members receive no payment for contributing to the application review process at scheduled meetings or for attending these meetings.
3.10 Is there any guidance tool available for making electronic applications? If yes, provide the link and/or step-by-step instructions.
Please refer to Section 3.7 above.
3.11 Does EC/IRB require any mock screens/screenshots of participant-facing material on the app? If yes, do these need to be submitted in the local language?
With the Medical Research Council (MRC), HRA provides an online tool that gives guidance on consent and the preparation of information for participants. HRA does not expect applicants to simply follow a template, so their guidance will help you design appropriate and proportionate information.
Informed consent process and the adequacy and completeness of research participant information:
- If applicable, review the provision of information to research participants about the purpose of the database, its procedures, potential risks, benefits, and alternatives, so that the individual understands this information and can make a voluntary decision whether to enroll and continue to participate.
- Do the terms of the informed consent as set out in the information sheets/consent forms suitably inform participants how their data will be used? (IRAS A26, A29-1, A29-2, PIS & Part B Section C if ALC and Part B Section 7 if children.)
- If applicable, is consent taken as part of a process with research participants having adequate time to consider the information, and the opportunity to ask questions?
- Is the language used clear and understandable to the research participants it is aimed at? If the Database will include children, has age-appropriate information been provided?
- Has informed consent already been given to use the data for research? If not, will consent now be sought? If informed consent has already been given for the use of the data in research, has evidence or an explanation of the terms of this consent been provided? Consider the requirements to seek consent from new participants, further consent from previous participants, or consent from relatives where the participants are deceased. (IRAS A26 and A28-A30)
- Suitably representative public involvement in the planning of the informed consent process (where applicable) can provide assurances about the points above.
3.12 Does the EC/IRB have any template or specific element requirements on ICF and/or other participant-facing materials?
Please refer to Section 7.7 of this guidebook on the DCT component – eConsent.
3.13 Is there any guidance tool on participant compensation including clinical study-related injury per local requirements?
With the Medical Research Council (MRC), HRA provides an online tool that gives guidance on consent and the preparation of information for participants. HRA does not expect applicants to simply follow a template, so their guidance will help sponsors design appropriate and proportionate information.
For all/most studies:
- The Participant Information Sheet (PIS) should describe how any complaints will be handled and what compensation may be available in the event of anyone being harmed. This information must be applicable to the setting in which the research will be conducted (e.g. university, NHS, commercial or other public research facility, etc.)
- Harm – You should provide potential participants with details of what redress and/or compensation should be available to them in the event that they are harmed as a consequence of taking part in your research. Details of insurance/indemnity schemes should be given, including whether compensation is dependent on demonstrating negligence or otherwise. If you are unsure what indemnity or insurance is available to you, you should speak to your R&D/research/research governance office.
- NHS-based research – NHS bodies are liable for clinical negligence and other negligent harm to individuals covered by their duty of care. NHS Institutions employing researchers are also liable for negligent harm caused by the design of studies they initiate. NHS Indemnity does not offer no-fault compensation (i.e. for non-negligent harm), but they may offer an ex gracia payment. If NHS indemnity is in place for your study, you could include the following possible wording: "In the event that something does go wrong and you are harmed during the research and this is due to someone's negligence, then you may have grounds for legal action for compensation against [name of Sponsor Organization, NHS Trust, Private Clinic] but you may have to pay your legal costs. The normal National Health Service complaints mechanisms will still be available to you (if appropriate)."
- Universities – Universities employing researchers are liable for their employees' actions (undertaken as part of their job) and are expected to insure against the risk of claims relating to research studies that their staff design and undertake. They may have insurance that covers both negligence and no-fault compensation; this would normally exclude clinical negligence for which NHS bodies are liable. Appropriate statements should be included in the Participant Information Sheet (PIS), which describes what insurance cover is being provided, in terms that a layperson would understand.
- Commercial research – For a Pharmaceutical industry-sponsored study, where there are Association of the British Pharmaceutical Industry (ABPI) or other no-fault compensation arrangements, you should include the following form of words in your PIS: "We will provide compensation for any injury caused by taking part in this study in accordance with the guidelines of the Association of the British Pharmaceutical Industry (ABPI). We will pay compensation where the injury probably resulted from A drug being tested or administered as part of the trial protocol or any test or procedure you received as part of the trial."
- Any payment would be without legal commitment and would not be bound by these guidelines to pay compensation where the injury resulted from a drug or procedure outside the trial protocol or where the protocol wasn't followed.
3.14 Are there any specific local safety reporting requirements for clinical studies?
The procedures for safety reporting will vary depending on your type of study. See the detailed response under Section 2.15 of this guidebook on safety reporting.
3.15 Does the EC/IRB require any periodic study reporting?
If the study was reviewed by a Research Ethics Committee (REC) in Scotland or Northern Ireland, an annual progress report should be submitted 12 months after the date on which the favorable ethics opinion was given. However, progress reports are not required in the following instances:
- If the study is expected to run for less than two years in duration.
- If the study received a proportionate review.
- If the study received a favorable ethics opinion from a REC in England or Wales.
If the study was given a favorable ethics opinion by a REC in Scotland or Northern Ireland, there are separate forms for submitting progress reports, depending on the type of research. Please use the form that is applicable to your type of research.
Forms should be completed in typescript. They will need to be authorized by the Chief Investigator or the sponsor/sponsor representative.
An electronic copy should be emailed to the REC within 30 days of the end of the reporting period.
- Annual progress report form for clinical trials of investigational medicinal products (CTIMPs)
- Annual progress report form for all other research
- Annual Report form for Research Databases
- Annual Report form for Research Tissue Banks
HRA and HCRW Approval
For research with HRA and HCRW Approval which were not required to be reviewed by a REC, progress reports are not required.
Confidentiality Advisory Group (CAG) annual review
All approvals are reviewed annually to assess the need for continuing approval and to ensure that progress towards, or achievement of, any conditions of approval is in place. This review is carried out following submission of an annual review report.
At this stage, you should consider if it would be possible to reduce the amount of confidential patient information that you are processing.
To allow sufficient time for processing, an annual review report should be submitted to the Confidentiality Advice Team by email four weeks before the approval expires (i.e. no later than 11 months following the final approval date) using the report template. This will be assessed by the Confidentiality Advice Team in the first instance.
Early termination or temporary halt of research
If the research is terminated early or is temporarily suspended, you should notify all relevant review bodies within 15 days.
Was this article helpful?