10. Roadmap of the Submission Process of a Clinical Trial
- 10 Mins to read
- DarkLight
10. Roadmap of the Submission Process of a Clinical Trial
- 10 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
10.1 Clinical Trial with Investigational Medicinal Product
Combined Review is the way to seek approvals for new Clinical Trials of Investigational Medicinal Products and combined medicine and device trials.
A single application using the IRAS goes to both the MHRA and the Research Ethics Committee (REC) at the same time. The application also goes for study-wide review, such as HRA and HCRW approval, if the study is to take place in the NHS or Northern Ireland HSC.
The regulatory and ethics reviews are done in parallel and any request for further information (RFIs) is raised jointly. A single response to these requests leads to a single decision from both reviews. Study-wide review is usually issued at the same time as MHRA and REC but may come later if there are still issues to discuss with the applicant.
IRAS is used for the initial application and supports the trial through the amendments right up to the end of the trial. Research teams can allocate different roles in the system for colleagues working on a project.
Initial review process and timelines
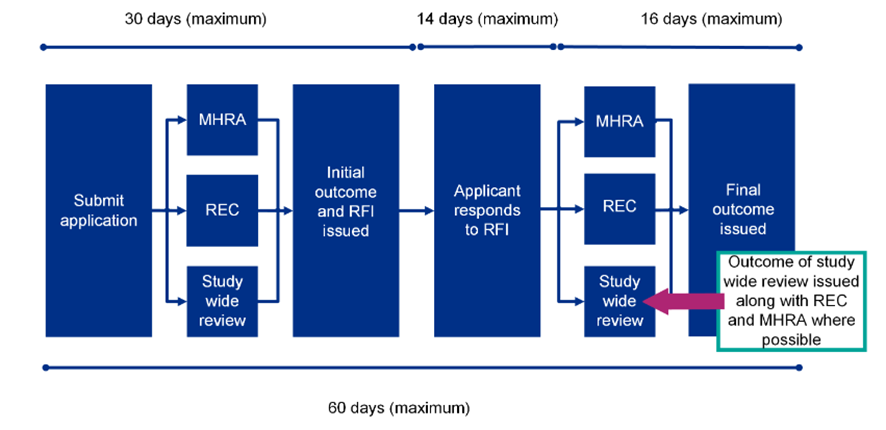 If there are questions during the review, a ‘Request for Further Information’ (RFI) may be raised, and applicants will need to provide additional information or clarifications. Applicants can request an extension if more than 14 days are needed to respond to the RFI, by emailing the MHRA at clintrialhelpline@mhra.gov.uk. If more than 14 days are needed, this will affect the 60-day timeline.
If there are questions during the review, a ‘Request for Further Information’ (RFI) may be raised, and applicants will need to provide additional information or clarifications. Applicants can request an extension if more than 14 days are needed to respond to the RFI, by emailing the MHRA at clintrialhelpline@mhra.gov.uk. If more than 14 days are needed, this will affect the 60-day timeline.
For help with tasks in the new IRAS, the Service Desk can be contacted at service.desk@hra.nhs.uk.
The step-by-step guide to using the IRAS for combined review provides a detailed description of the process. It is advisable to follow the webpage to ensure that the most up-to-date information and instructions are observed.
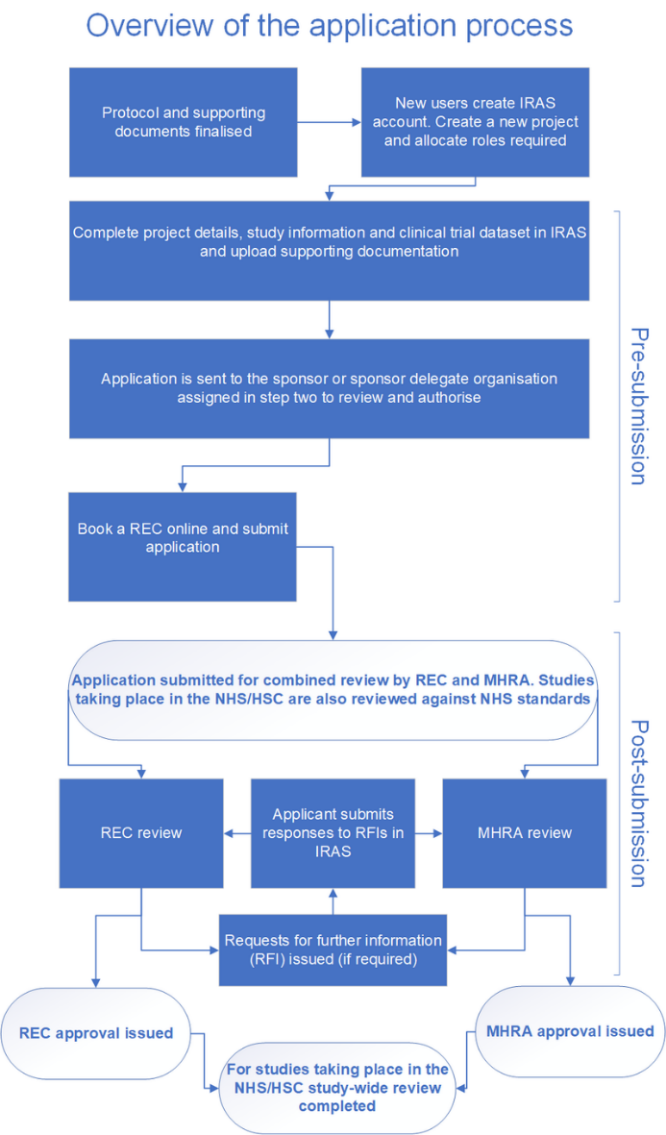
The flowchart below shows how the combined review process works and the steps to submit an application.
Visual graphic describing the overview of the application process
Figure 1: Overview of the application process
Contents
Overview of the application process
- Preparing your application
- Deleting a project
- Applications involving the use of ionising radiation or a medical device
- Validation
- Responding to Requests for Further Information
- Resending an application
- Withdrawing a submission
- Resubmitting a withdrawn application (or resubmitting an application that was not authorised by the regulator)
- Downloading documents
- Development Safety Update Reports (DSURs)
- Urgent Safety Measures (USMs)
- Notifying End of research and final report
10.2 Pediatric Investigation Plan (PIP)
Pediatric studies are part of the normal lifecycle and development of a medicine. Sponsors need to agree and carry out a PIP if they are applying for marketing authorization for a new medicine or, in certain cases, if they are developing a currently licensed medicine unless they have a waiver for all or a subset of the pediatric population. A PIP includes all the measures that would generate the necessary data from studies in children as well as measures with regard to formulation development.
Sponsors must submit a PIP or waiver application via MHRA Submissions.
The MHRA has published guidance on the submission and assessment of UK Pediatric procedures. These are:
- Procedures for UK Pediatric Investigation Plans
- The format and content of applications for agreement or modification of a Pediatric Investigation Plan and requests for waivers or deferrals and concerning the operation of the compliance check.
For general inquiries about pediatric submissions including PIP and waiver applications, modification procedures, and compliance checks, contact the MHRA Pediatric Unit at ukpip@mhra.gov.uk.
The PIP application process for applicants is simplified by offering an expedited assessment where possible, and by mirroring the submission format, content, and terminology of the EU-PIP system.
The legal requirements for UK-PIPs are set out in the Human Medicines Regulations 2012, as amended by the Human Medicines Regulations (Amendment etc.) (EU Exit) Regulations 2019 (HMRs), including transitional provisions.
PIP Submissions
- UK PIP submissions after 01 January 2021
The MHRA is accepting PIP applications submitted via the MHRA Submissions portal.
According to Regulation 50B (3) of the Human Medicines Regulations 2012, as amended by the Human Medicines (Amendment etc.) (EU Exit) Regulations 2019 (HMRs), including transitional provisions, applications for the agreement of a pediatric investigation plan should be submitted, unless duly justified, ‘not later than upon completion of the human pharmacokinetic (PK) studies’, as specified in section 5.2.3 of Part I of Annex I to the 2001 Directive,’ unless the MHRA agrees to accept a later request.
For details on the application, please read the published guidance on the required format and content.
When a PIP application is submitted to MHRA, information should be provided on whether there is:
- an agreed EU-PIP and the opinion and supporting documentation are included;
- an ongoing EU-PIP assessment and its timeline in the PDCO assessment cycle (i.e. day 30, 60, clock stop, day 90, or 120); or
- any current scientific divergence between the submitted PIP application and the EU-PIP.
This information will be used to establish the MHRA assessment process as described in Sections 1.3 to 1.4 below.
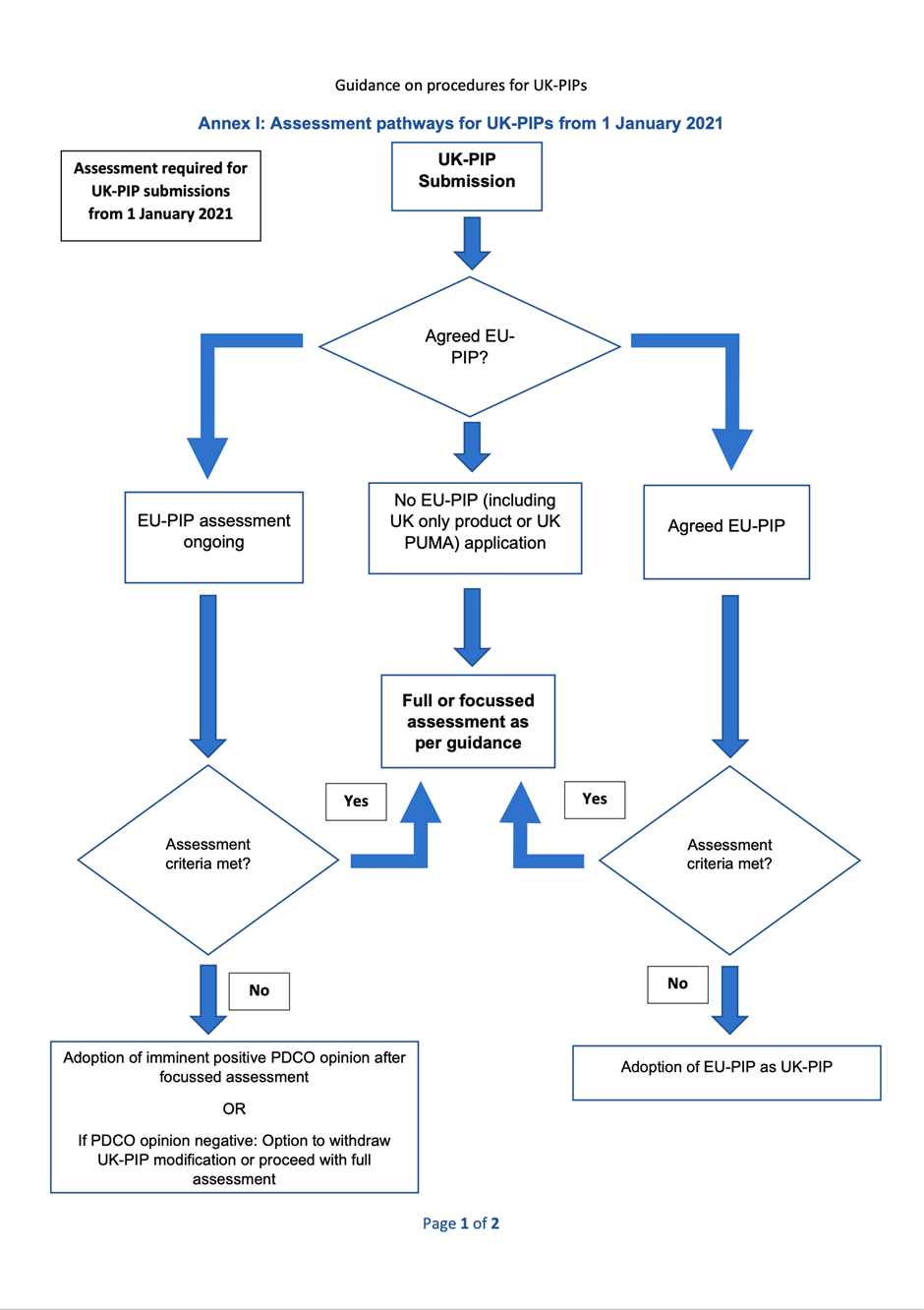
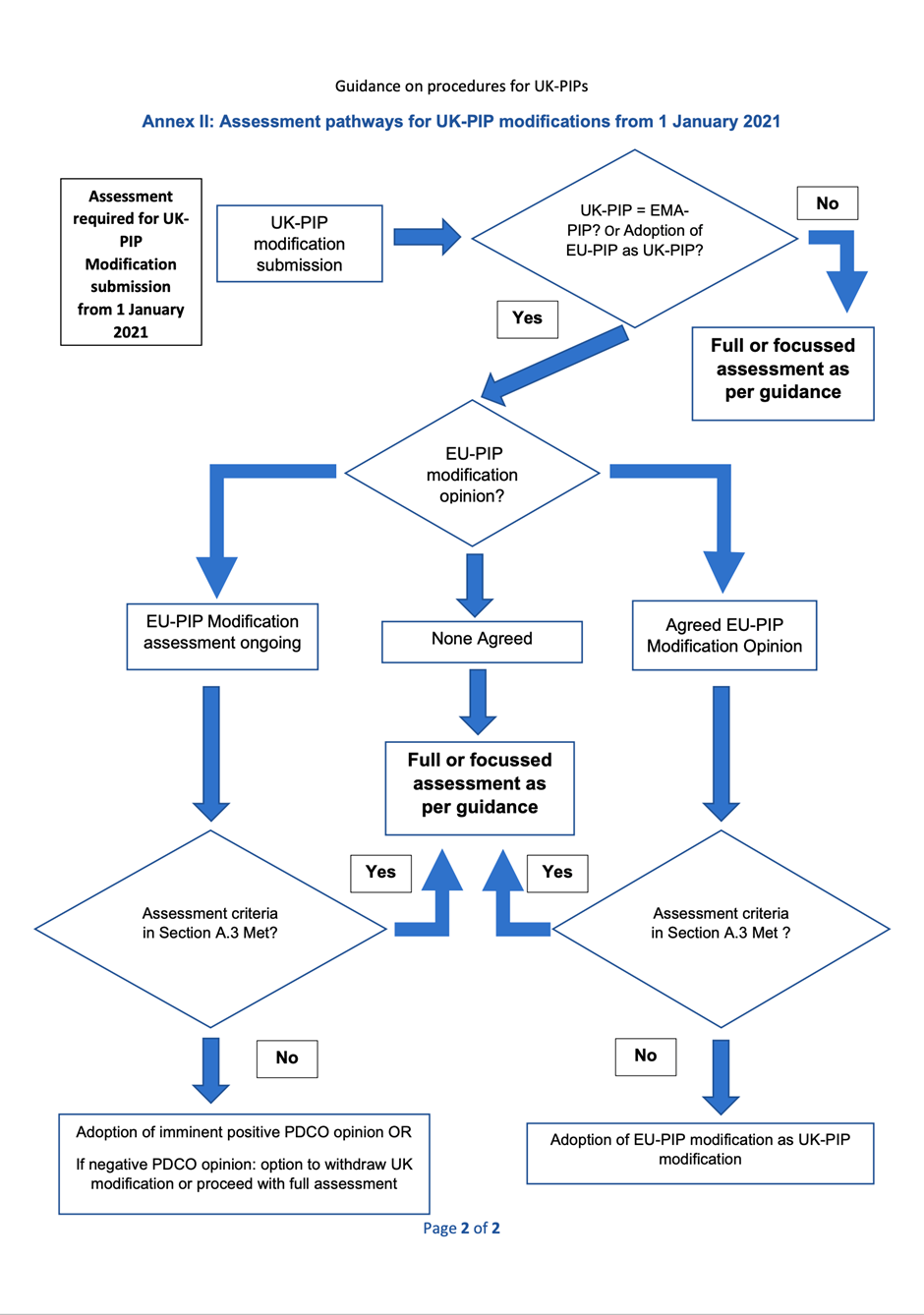
The assessment pathways for UK-PIP submissions are from Annexes I and II (PDF, 90.9 KB, 2 pages).
1.2 PIP submission with agreed PDCO opinion from 01 January 2021 or ongoing assessment at EMA from 01 January 2021:
In special circumstances when there is a delay in the PIP submission to the UK, in principle, the MHRA will aim to maintain alignment with a positive PDCO opinion, if one is reached before the MHRA assessment is completed.
However, divergence could occur as we will make decisions for PIPs based on national and NHS pediatric public health needs.
- Unmet UK pediatric needs [Section 6 of this guidance]
- Pediatric-only development particularly for an innovative product (such as a new drug class or mechanism of action)
- The incidence of the disease in the UK population
- The relevance of the scientific arguments by EMA / PDCO in the summary report (SR) to the UK pediatric population
- Any additional safety or efficacy concerns for the UK population
- The nature and number of licensed products already available for the intended pediatric indication
- The feasibility of performing the proposed pediatric studies in the UK only
- PIP is to support a UK Pediatric Use Marketing Authorization (PUMA)
A full assessment may be requested by the applicant.
1.3. UK-PIP with no EU-PIP from 1 January 2021
This section applies if there is no corresponding EU PIP submission or if the PDCO opinion is negative. A full assessment of the UK-PIP is required.
The applicant should additionally clarify if:
- there has been a previous negative EMA / PDCO PIP opinion,
- there was a withdrawn EU-PIP prior to the adoption of an EMA/PDCO opinion,
- the current UK submission has been updated since the previous negative or withdrawn EU-PIP,
- the applicant has included the previously withdrawn or negative EMA / PDCO PIP SR as part of the supporting documents in the MHRA submission, or
- during the assessment, consideration will be given to the scientific discussions of the EMA / PDCO which led to the negative opinion or the withdrawal of the EU-PIP.
If the applicant chooses to submit to the MHRA and has a negative PDCO opinion, the applicant should consider incorporating changes to the UK-PIP, for the elements that received a negative PDCO assessment.
- PIP Modifications
2.1. Modification of an adopted or agreed UK-PIP
According to Regulation 50B (6) of the UK’s Human Medicines Regulations 2012, as amended by the Human Medicines (Amendment etc.) (EU Exit) Regulations 2019 (HMRs), including transitional provisions, the applicant may request a modification of an agreed UK PIP if they encounter difficulties with its implementation as to render the plan unworkable or no longer appropriate.
The MHRA will assess the proposed changes based on the scientific arguments provided.
For both adopted and agreed UK-PIPs, when a PIP modification is submitted, it should be confirmed if there is:
- a modification submitted for an adopted or agreed UK-PIP,
- an agreed EU-PIP modification,
- an EU-PIP modification assessment ongoing, or
- a significant scientific divergence between the current agreed EU-PIP and the agreed UK-PIP.
Modifications submitted for UK-PIPs where there is an EU-PIP should include the most recent PDCO opinion and PIP SR.
For Agreed UK-PIPs, there will be either a focused assessment in cases where the EU opinion for the initial UK-PIP was accepted by the MHRA or a full modification assessment in cases where the initial UK-PIP underwent full assessment.
The applicant may request a full assessment.
2.2. Requests for a PIP modification from 1 January 2021
In principle, the MHRA will aim to accept a positive PDCO opinion on modifications in cases where the initial UK-PIP was agreed on the basis of an agreed EU-PIP. A focused assessment may be needed if the criteria in Section 1.3 are met.
If the PDCO opinion is negative whilst the UK assessment is ongoing:
- The applicant has the option to withdraw the UK-PIP modification request or continue with the MHRA assessment.
- Once a PIP has been withdrawn, a new UK-PIP modification can be submitted and will undergo review using the same assessment criteria discussed in Section 1.3 of this guidance.
- If continuing with the MHRA assessment, the applicant can discuss amendments to the proposals before the final MHRA opinion on the proposed Modification is agreed.
2.3. PIP modification with no agreed EU PIP modification
If there is no agreed EMA modification opinion, a full assessment of modification will be required.
- Waivers
3.1. Submission of Pediatric Class Waiver
The current EMA class waivers list will be adopted by the UK from 1 January 2021.
In principle, the MHRA will aim to accept a positive EMA opinion on a class waiver request. Where there is no EMA opinion, an MHRA assessment will be undertaken.
For a negative EMA opinion on whether a Class Waiver applies:
- The applicant should submit a full product-specific waiver request for MHRA assessment which should include EMA opinion on the class waiver.
- If there is an EMA opinion on the applicant’s subsequent product-specific waiver request, then this should be made available to determine if a focused or full assessment is required.
3.2. Submission of full product-specific waiver
Sections 1.2 to 1.4 also apply to MHRA submissions for full product-specific waivers.
For an EU full product-specific waiver with a positive PDCO opinion or EMA decision before 1 January 2021:
- These will be adopted as a UK full waiver. No submission to the UK is required.
- Compliance Check
4.1. Adopted UK-PIP Compliance check (CC)
According to Regulation 50A (3) of the UK’s Human Medicines Regulations 2012, as amended by the Human Medicines (Amendment etc.) (EU Exit) Regulations 2019 (HMRs), including transitional provisions, applicants must submit a CC application to the MHRA where one is required for validation of the UK MA application.
A positive PDCO CC or interim CC will be adopted as the UK CC outcome unless subsequent modifications have led to a divergence between the UK- and EU-PIPs.
However, the applicant must pay particular attention to the agreed timelines of those measures which would need to be completed after the PDCO CC to ensure compliance on the date of the UK Marketing Authorization (MA) submission.
The PDCO compliance outcome documents should be submitted ahead of, or at the time of MA application via the PIP portal of MHRA Submissions (user guides available via the PIP tile). The format and submission procedure for PIP applications to the MHRA are published separately.
At the end of the CC procedure, an MHRA compliance report will be issued to the applicant with either an outcome letter (for partial CC) or a decision (final or full CC).
4.2. Agreed UK-PIP CC
According to Regulation 50A (3) of the UK’s Human Medicines Regulations 2012, as amended by the Human Medicines (Amendment etc.) (EU Exit) Regulations 2019 (HMRs), including transitional provisions, applicants must submit a CC application to the MHRA where one is required for validation of the UK MA application.
A UK assessment is required for full or interim CC if:
- there is any scientific divergence between the agreed UK-PIP and the EU-PIP,
- there is no PDCO CC, or
- in the case of an interim CC, there are additional key measures completed prior to UK MA submission.
The MHRA will adopt the PDCO CC outcome if:
- there is a positive PDCO CC,
- the UK-PIP is equivalent to the EU-PIP, or
- for interim CC, there are no additional key measures completed prior to UK MA submission that require further CC assessment.
Applicants are encouraged to request a CC ahead of submission of an MA application where one is required for validation. This should be submitted via the PIP portal of MHRA Submissions. At the completion of the CC procedure, the MHRA will issue compliance outcome documents similar to those noted in Section 4.1.
4.3. Non-Compliance
- For non-compliance due to (minor) administrative issues, or discrepancies that do not affect the scientific conduct of the study, a streamlined assessment will be proposed at the time the applicant is informed of the noncompliance outcome.
- This streamlined assessment will combine a shortened modification procedure with a rapid CC.
- If the above is not applicable, the applicant will be required to submit a modification for a full assessment to align the non-compliant key elements of the opinion with those of the completed study report.
- A rapid CC will be offered at the end of a positive modification agreement.
4.4. Statements of compliance
An MHRA statement of compliance when all of the agreed PIP measures have been completed, will be issued, if appropriate, when an MA application (initial, extension, or variation) is granted:
“The development of this product has complied with all measures in the agreed pediatric investigation plan < reference number >.”
The Summary of Product Characteristics and, where applicable, the package leaflet will include the results of the studies referred to in the UK-PIP.
- Pediatric Study Plans (PSP)
5.1. Pediatric study plans (PSP)
Regarding the applicant’s pediatric study plans (PSP) agreed by the US Food and Drug Administration (FDA), applicants should provide the agreed PSP as part of their UK-PIP submission.
- Unmet needs in the UK pediatric population
The unmet needs of the UK pediatric population will be defined by:
- Therapeutic areas identified by UK health bodies as high-priority public health concerns.
- Product development in conditions identified after consultations with UK experts and patient groups, including those for rare diseases identified under the auspices of the Department of Health and Social Care (DHSC) policy paper - UK strategy for rare diseases.
- Product development in conditions (or pediatric groups) identified as critically important in the Pediatric Regulation 10-year report.
- Products that are intended to be authorized as orphan medicines.
- Products that fulfill the criteria of promising/innovative new products and are part of an accelerated MHRA submission or assessment pathway.
10.3 Clinical Study (Interventional or Observational)
To assist organizations in determining whether a project is research and the level of approval required, the HRA provides different tools.
Was this article helpful?