10. Roadmap of the Submission Process of a Clinical Trial
- 4 Mins to read
- DarkLight
10. Roadmap of the Submission Process of a Clinical Trial
- 4 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
10.1 Clinical Trial with Investigational Medicinal Product
A trial application must be made via CTIS.
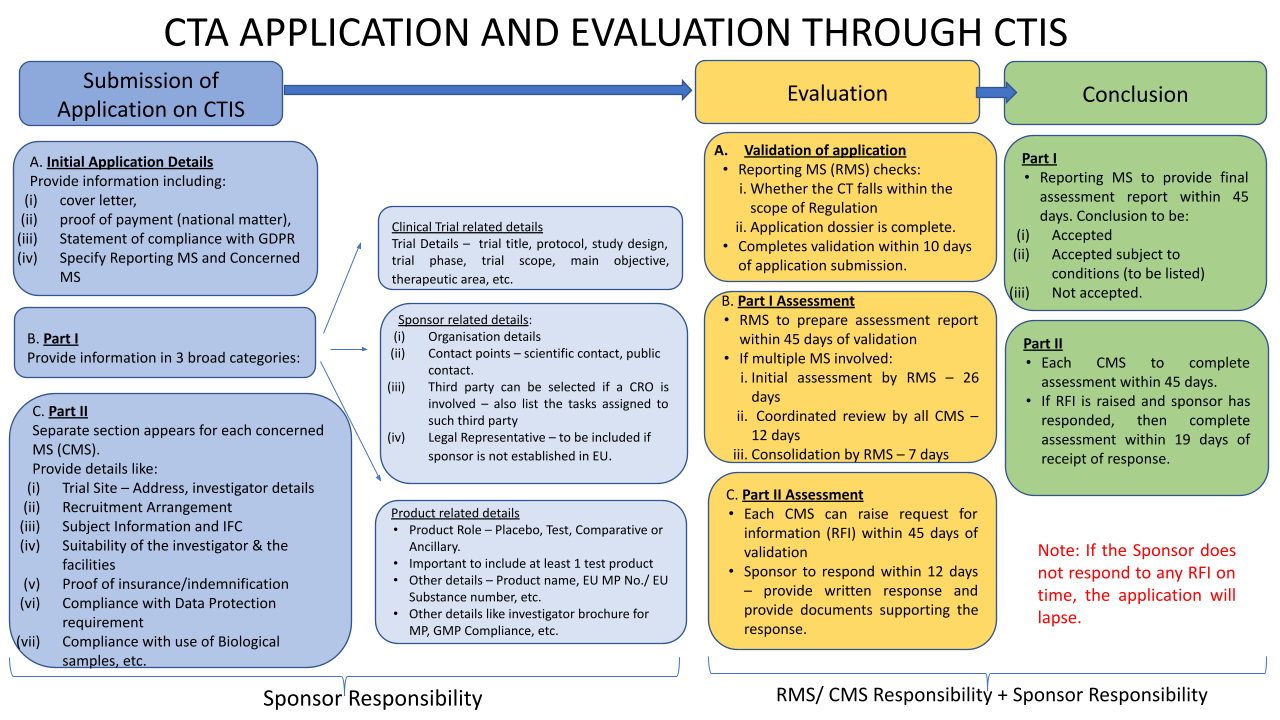
The BfArM states that:
“CTIS will be the single entry point for submitting clinical trial information in the EU and the European Economic Area (EEA). CTIS will contain a Sponsor workspace for clinical trial sponsors and the organizations that work with them, an Authority workspace for EU Member States, EEA countries, and the European Commission, and a public website.
The Sponsor secure workspace will assist clinical trial sponsors in preparing and compiling the clinical trial application and related documents to submit for assessment.
The Authority secure workspace will support the activities of EU Member States, EEA countries, and the European Commission in assessing and overseeing clinical trials.
Through the public website, members of the public can access detailed information on all clinical trials conducted in the EU and the EEA once trials start to be submitted and approved in CTIS.
EU Member States and EEA countries will assess and supervise clinical trials in CTIS, while EMA sets up and maintains CTIS. The European Commission ensures the correct interpretation and implementation of the Clinical Trials Regulation.”
Information in a Q&A format, together with guidance on how to prepare for CTIS, is available on the BfArM website.
10.2 Pediatric Investigation Plan (PIP)
A pediatric investigation plan (PIP) is a development plan aimed at ensuring that the necessary data are obtained through studies in children, to support the authorization of a medicine for children. All applications for marketing authorization for new medicines have to include the results of studies as described in an agreed PIP unless the medicine is exempt because of a deferral or waiver.
This requirement also applies when a marketing authorization holder wants to add a new indication, pharmaceutical form, or route of administration for a medicine that is already authorized and covered by intellectual property rights.
Deferrals and Waivers
The Pediatric Committee (“PDCO”) may grant PIP deferrals for some medicines. These allow an applicant to delay the development of the medicine in children until, for instance, there is enough information to demonstrate its effectiveness and safety in adults. Even when studies are deferred, the PIP will include details of the pediatric studies and their timelines.
Reporting under Deferrals
A. Annual Report
Marketing authorization holders who have received a deferral on a PIP are obliged to submit annual reports to the EMA. These reports should provide an update on progress with pediatric studies in accordance with the decision of the EMA agreeing on the PIP and granting a deferral.
- New medicines
If an agreed PIP for a new medicine contains a deferral, the applicant needs to send the first annual report to the Agency in the month before the first anniversary of the date of marketing authorization. There is no need to submit annual reports before the marketing authorization is granted.
- Medicines already authorized
- if the PIP decision is agreed upon less than six months before the anniversary date of the first marketing authorization granted in the European Economic Area, the marketing authorization holder should send the annual report in the month before the next anniversary of the date of the marketing authorization;
- if the anniversary date occurs more than six months after the date of the PIP decision, the report should be sent in the month before the anniversary date.
Use the date of the decision on the initially agreed PIP to calculate the due date of the first annual report. Do not use the date of any subsequent modifications of the agreed PIP.
B. Subsequent Reports
Subsequent annual reports need to be submitted every year, within the sixty days preceding the anniversary of the marketing authorization date.
The PDCO may also grant waivers when the development of a medicine in children is not needed or is not appropriate, such as for diseases that only affect the adult population.
Post Assessment
After assessing an application for a PIP, deferral, waiver, or modification, the PDCO formulates an opinion, which is notified to the applicant. The applicant is then able to request a re-examination of the opinion if s/he wishes. Once the Committee has issued its final opinion, after re-examination if requested, the EMA adopts a decision.
The EMA makes all opinions and decisions on PIPs, deferrals, and waivers public, after deletion of information of a commercially confidential nature.
Scientific Advice
Applicants can request scientific advice from EMA in preparation for a PIP, which is free of charge for questions relating to the development of pediatric medicines. They can also follow up a PIP with scientific advice, for example on combined adult and pediatric development in light of the PIP requirements.
EMA discourages applicants from submitting scientific advice and a PIP application in parallel.
For further information, please see the PIP-related page by the EMA.
10.3 Clinical Study (Interventional or Observational)
Section 4(23) of the German Medicinal Products Act states that the provisions that are applicable to clinical trials do not apply to an investigation that is a non-interventional trial.
(23) A clinical trial is one within the meaning of point (2) of Article 2(2) of Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on clinical trials on medicinal products for human use and repealing Directive 2001/20/EC. OJ L 158, 27.5.2014, p. 1; (OJ L 311, 17.11.2016, p. 25). No clinical trial is a non-interventional study within the meaning of point (4) of Article 2(2) of Regulation (EU) No 536/2014.
BfArM provides information on non-interventional studies.
Was this article helpful?