10. Roadmap of the Submission Process of a Clinical Trial
- 4 Mins to read
- DarkLight
10. Roadmap of the Submission Process of a Clinical Trial
- 4 Mins to read
- DarkLight
Article summary
Did you find this summary helpful?
Thank you for your feedback
10.1 Clinical Trial with Investigational Medicinal Product
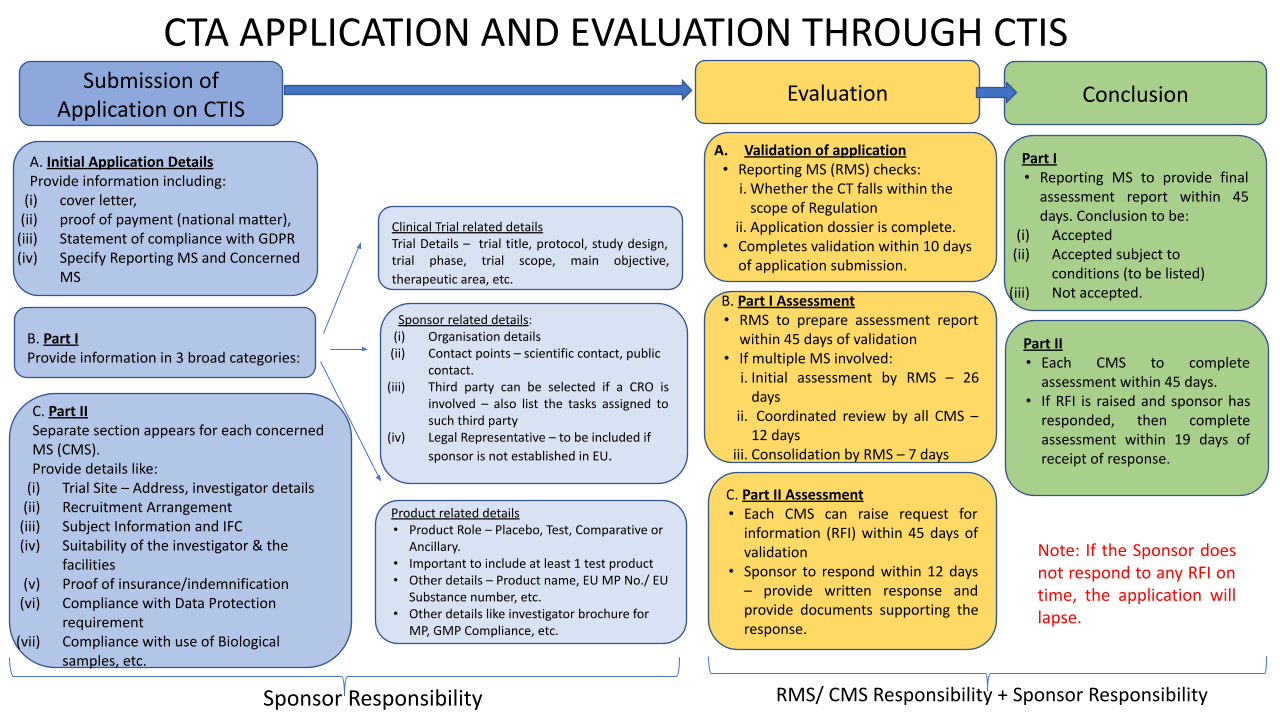
The submission process using CTIS (see Annex I for details on how to get started using the CTIS itself) is described in more detail below this flowchart/roadmap:
The EU's new Clinical Trial Regulation (EU) 536/2014 (“EU CTR”) came into force on 31st January 2022.
The aim of the EU CTR is to ensure a greater level of harmonization of the rules of conducting clinical trials throughout the EU. Subject to some transitional provisions, the EU CTR repeals the EU’s Clinical Trials Directive 2001/20/EC. The EU CTR features:
- A streamlined application procedure for all clinical trials conducted in Europe via a single EU portal and database. All applicants must be registered before submission of a clinical trial application.
- A single authorization procedure for all clinical trials, to allow a faster and more thorough assessment by all concerned EU countries.
- The extension of the silent agreement principle to the authorization process (After validation of the application (10 to 25 days), the assessment of Part I should last 45 days. The assessment for Part II, which runs in parallel, should also last 45 days. Then, each Member State Concerned has five days to give its decision on whether the trial may proceed, if a Member State does not provide its decision within the five-day timeframe, it will be understood that it has tacitly approved the assessment of Part I and that will conclude the Part I assessment report) giving more legal certainty to sponsors and researchers, in particular SMEs and academics.
- Strengthened transparency for clinical trials data - the EU CTR aims to increase the availability of information on clinical trials through the EU clinical trial portal and database. Art 81(4) of the EU CTR states that the (information in the) EU database shall be publicly available unless confidentiality is justified, (e.g., in order to protect personal data or commercially confidential information). The CTIS serves as a key instrument to increase the transparency of clinical trials by offering searchable clinical trial information to patients, healthcare professionals, and the general public. Clinical trial results will be available as a technical summary and in lay language.
Implementation of the EU CTR is by way of a phased transition that will happen over a 3-year period, whereby, during that period, organizations who have clinical trials running, will progressively transfer ongoing clinical trials over to the new system in accordance with the rules of the EU CTR, at the latest by 31st January 2025. From 31st January 2023, all new applications for clinical trials must be registered using the new system established in the EU CTR.
The previous Clinical Trials Directive 2001/20/EC ('Clinical Trials Directive') is repealed with effect from 31st January 2022. The Clinical Trials Directive will, however, continue to apply until 31st January 2025 in respect of:
- Clinical trial applications submitted before 31st January 2022; and
- Clinical trial applications submitted before 31st January 2023, if the sponsor opted to submit in accordance with the Clinical Trials Directive (rather than via the EU CTR’s CTIS portal).
During the transitionary period EU Member States may continue to apply their national law, adopted to transpose Directive 2003/94/EC to those clinical trials conducted under the Clinical Trials Directive. In other words, EU Member States may continue to apply the current regulatory regime until 31st January 2025.
Submission of applications – Process
Under the new EU CTR, applications for trials with medicinal products in humans can be submitted through a new Clinical Trial Information System (CTIS) - accessible via euclinicaltrials.eu - along with amendments and other changes to ongoing clinical trials.
The CTIS includes workspaces for clinical trial sponsors and authorities as well as a public website. In order to access the CTIS Sponsor workspace, users need an active EMA account. Further, the users need to register their organization and trials via the Organization Management Service (OMS) from which CTIS retrieves its data. In addition, before completing the clinical trial application in CTIS, the sponsors should ensure that the details of the medicinal products used in the clinical trials are already registered in the eXtended EudraVigilance Medicinal Product Dictionary (xEVMPD). A placebo can be added manually in CTIS directly. Further, the active substance for the developed medicinal product must be available in EMA SMS (Substance Management Service).
Submission & Choice of Reporting Member State
The application is submitted via the CTIS portal by the Sponsor and will include details for all Concerned Member States (CMS) where the Sponsor intends to conduct the clinical trial.
The Sponsor will nominate a Reporting Member State who will act as the single point of contact with regard to the authorization process.
In terms of choosing the Reporting Member State, if the proposed Clinical Trial is deemed to be a low-intervention clinical trial (Art.2.2 (3) EU CTR (e.g., the medicinal product has marketing authorization in one of the CMS, or it is an investigational medicinal product which is to be used in the Clinical Trial in accordance with published scientific data in relation to its safety and efficacy in one of the CMS), then the Member State to which this criteria applies should be nominated as the Reporting Member State.
Apart from these criteria, the EU CTR is silent as to the choice of the Reporting Member State. However, the Reporting Member State nomination may not always be granted, and it is on Day 6 following submission that the Reporting Member State is confirmed.
More information can be found in the EMA CTIS - Sponsor Quick Guide.
10.2 Pediatric Investigation Plan (PIP)
A Pediatric Investigation Plan (PIP) is a development plan aimed at ensuring that the necessary data are obtained through studies in children, to support the authorization of a medicine for children. All applications for marketing authorization for new medicines have to include the results of studies as described in an agreed PIP unless the medicine is exempt because of a deferral or waiver.
EMA information on the Pediatric Investigation Plan can be found here.
10.3 Clinical Study (Interventional or Observational)
The EU CTR does not apply to non-interventional studies.
Was this article helpful?